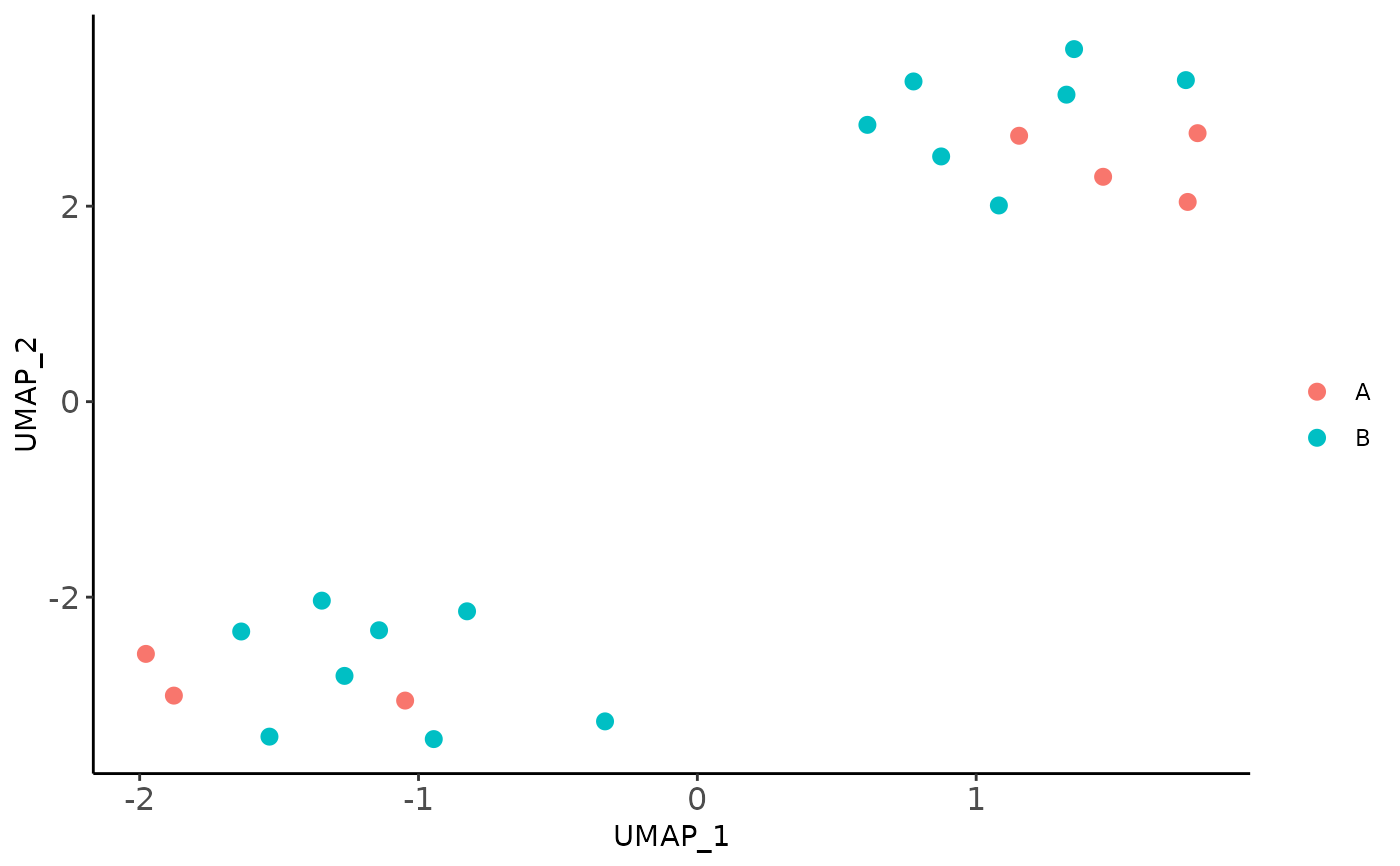
Visualize sample variability in a 2D space
plot_sample_2D.RdPerforms dimensionality reduction of factor scores for the visualization of sample-level variability
Usage
plot_sample_2D(
model,
group = FALSE,
method = "UMAP",
metadata,
sample_id_column = "sample",
color_by,
...
)Arguments
- model
A MOFA2 model.
- group
Boolean flag TRUE/FALSE, to specify if a grouped MOFA model is provided.
- method
A string specifying if "UMAP" or "MDS" should be performed
- metadata
A data frame containing the annotations of the samples included in the MOFA model.
- sample_id_column
A string character that refers to the column in
metadatawhere the sample identifier is located.- color_by
A string character that refers to the column in
metadatawhere the covariate to be tested is located.- ...
inherited parameters of
uwot::umap()
Details
For a MOFA2 model it performs a multidimensional scaling (MDS)
or uniform manifold approximation and projection (UMAP) of the factor
scores. It allows you to color samples based on a covariate available in
your meta-data.
Examples
inputs_dir <- base::system.file("extdata", package = "MOFAcellulaR")
model <- MOFA2::load_model(file.path(inputs_dir, "testmodel.hdf5"))
#> Warning: Factor(s) 1 are strongly correlated with the total number of expressed features for at least one of your omics. Such factors appear when there are differences in the total 'levels' between your samples, *sometimes* because of poor normalisation in the preprocessing steps.
metadata <- readRDS(file.path(inputs_dir, "testmetadata.rds"))
UMAP_embedding <- plot_sample_2D(model = model,
group = FALSE,
method = "UMAP",
metadata = metadata,
sample_id_column = "sample",
color_by = "batch")