Overview
There are many methods that allow us to extract biological activities from omics data. decoupleR is a Bioconductor package containing different statistical methods to extract biological signatures from prior knowledge within a unified framework. Additionally, it incorporates methods that take into account the sign and weight of network interactions. decoupleR can be used with any omic, as long as its features can be linked to a biological process based on prior knowledge. For example, in transcriptomics gene sets regulated by a transcription factor, or in phospho-proteomics phosphosites that are targeted by a kinase. This is the R version, for its faster and memory efficient Python implementation go here.
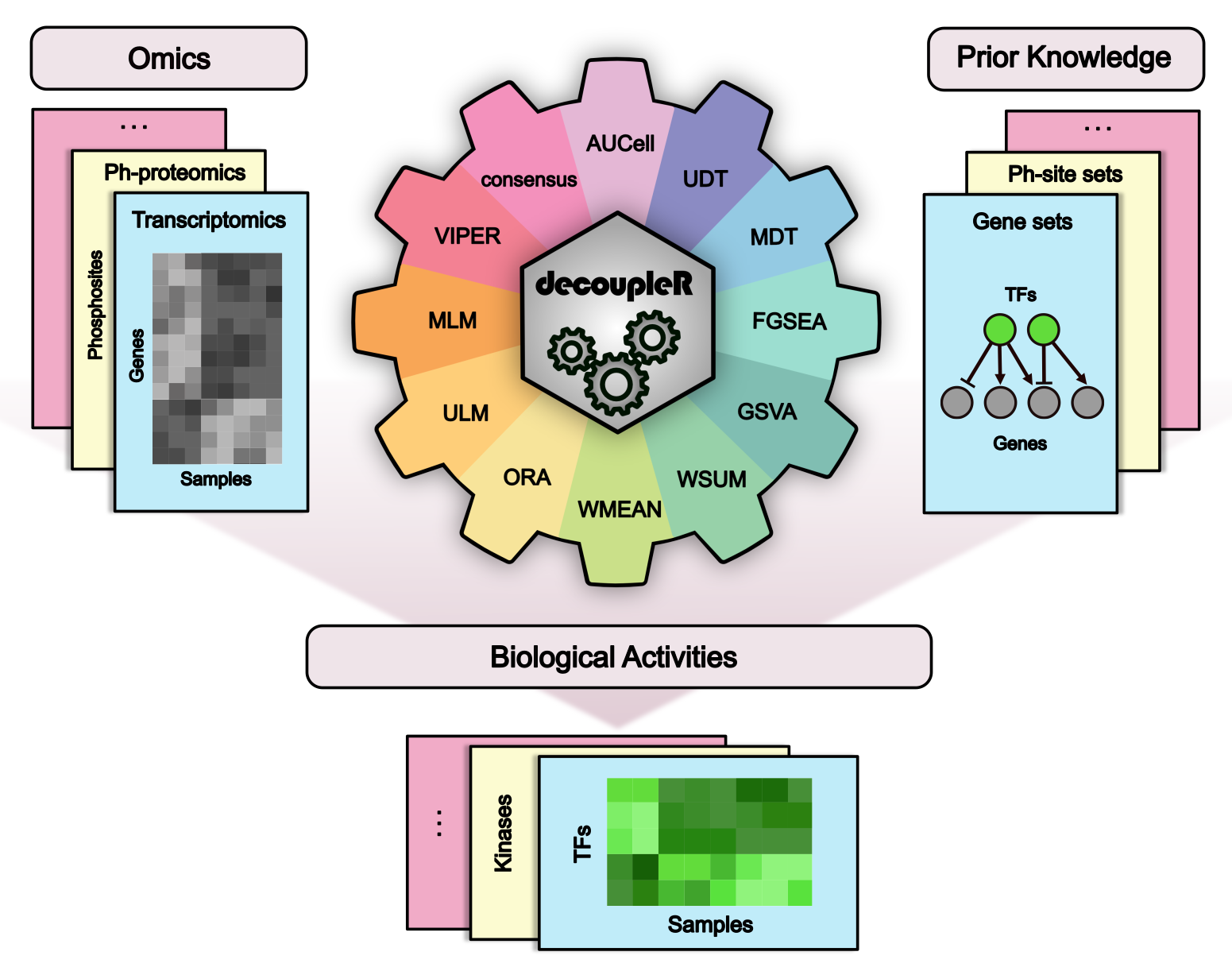
For more information about how this package has been used with real data, please check the following links:
- decoupleR’s general usage
- Pathway activity inference in bulk RNA-seq
- Pathway activity inference from scRNA-seq
- Transcription factor activity inference in bulk RNA-seq
- Transcription factor activity inference from scRNA-seq
- Example of Kinase and TF activity estimation
- decoupleR’s manuscript repository
- Python implementation
Installation
decoupleR is an R package distributed as part of the Bioconductor project. To install the package, start R and enter:
install.packages('BiocManager')
BiocManager::install('saezlab/decoupleR')Alternatively, if you find any error, try to install the latest version from GitHub:
install.packages('remotes')
remotes::install_github('saezlab/decoupleR')License
Footprint methods inside decoupleR can be used for academic or commercial purposes, except viper which holds a non-commercial license.
The data redistributed by OmniPath does not have a license, each original resource carries their own. Here one can find the license information of all the resources in OmniPath.
Citation
Badia-i-Mompel P., Vélez Santiago J., Braunger J., Geiss C., Dimitrov D., Müller-Dott S., Taus P., Dugourd A., Holland C.H., Ramirez Flores R.O. and Saez-Rodriguez J. 2022. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinformatics Advances. https://doi.org/10.1093/bioadv/vbac016