## Overview
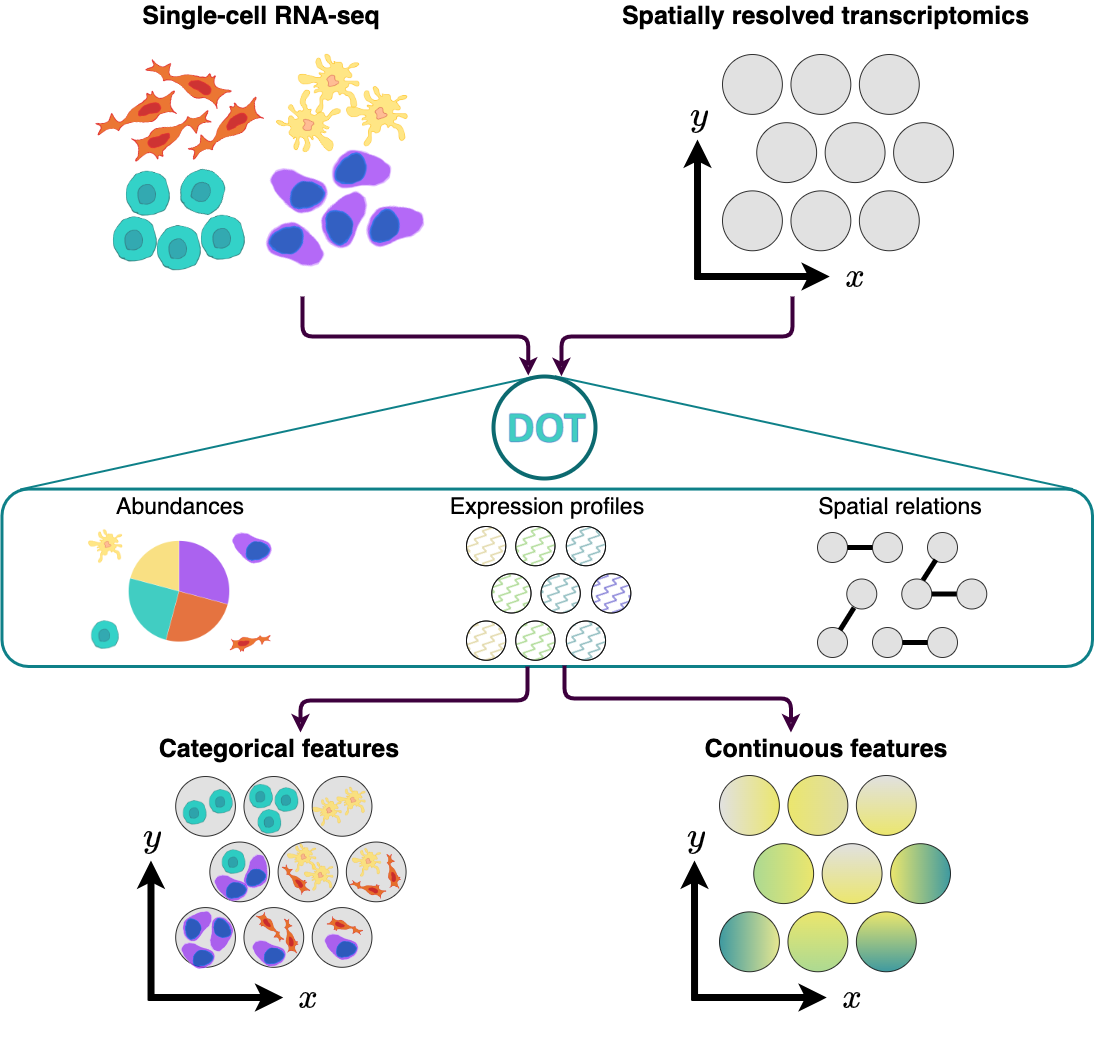
DOT is a method for transferring cell features from a reference single-cell RNA-seq data to spots/cells in spatial omics. It operates by optimizing a combination of multiple objectives using a Frank-Wolfe algorithm to produce a high quality transfer. Apart from transferring cell types/states to spatial omics, DOT can be used for transferring other relevant categorical or continuous features from one set of omics to another, such as estimating the expression of missinng genes or transferring transcription factor/pathway activities.
For more information about how this package has been used with real data and expected outputs, please check the following link:
Installation
DOT is available under the R package DOTr which you can install from GitHub with:
devtools::install_github("saezlab/DOT")Dependencies
- R (>= 4.0)
- R packages: fields, ggplot2, Matrix, methods, Seurat, stats, stringr
For optimal performance on moderately sized instances, we recommend at least 4 GB of RAM. For large reference scRNA-seq data or very large spatial instances higher memory may be required.
Installation takes less than five minutes. The sample dataset provided can be run in less than a minute on a “normal” desktop computer. DOT takes approximately 7 minutes to process a MERFISH MOp dataset with approximately 250 genes, 100 cell types and 4,000 spots.
Operating system tested on: macOS Monterey 12.4
Python version
Erick Armignol (https://github.com/earmingol) has implemented a native Python version of DOT, supporting also deployment on GPUs.
Check out https://github.com/earmingol/DOTpy for information on how to install it, examples of use and more practical tips.
Thank you Erick!
Citation
If you use DOT for your research please cite the following article:
Rahimi, A., Vale-Silva, L.A., Fälth Savitski, M. et al. DOT: a flexible multi-objective optimization framework for transferring features across single-cell and spatial omics. Nat Commun 15, 4994 (2024). https://doi.org/10.1038/s41467-024-48868-z.