PROGENy pathway signatures: Application to Bulk transcriptomics
Michael Schubert
2021-08-11
Source:vignettes/progenyBulk.Rmd
progenyBulk.RmdPROGENy pathway signatures
This R package provides the model we inferred in the publication "Perturbation-response genes reveal signaling footprints in cancer gene expression" and a function to obtain pathway scores from a gene expression matrix. It is available on bioRxiv.
Scoring the airway package data for pathway scores
This is to outline how to prepare expression data, in this case from the airway package for pathway activity analysis using PROGENy.
Preparing the gene expression matrix
library(airway)
library(DESeq2)
data(airway)
# import data to DESeq2 and variance stabilize
dset = DESeqDataSetFromMatrix(assay(airway),
colData=as.data.frame(colData(airway)), design=~dex)
dset = estimateSizeFactors(dset)
dset = estimateDispersions(dset)
gene_expr = getVarianceStabilizedData(dset)
# annotate matrix with HGNC symbols
library(biomaRt)
mart = useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
genes = getBM(attributes = c("ensembl_gene_id","hgnc_symbol"),
values=rownames(gene_expr), mart=mart)
matched = match(rownames(gene_expr), genes$ensembl_gene_id)
rownames(gene_expr) = genes$hgnc_symbol[matched]Obtaining pathway scores
We can then use the progeny function to score the expression matrix. Note that we are scaling the pathway scores with respect to the controls only.
Checking for differences between the groups
So now we might be interested how the treatment with dexamethasone affects signaling pathways. To do this, we check if the control is different to the perturbed condition using a linear model:
library(dplyr)
result = apply(pathways, 1, function(x) {
broom::tidy(lm(x ~ !controls)) %>%
filter(term == "!controlsTRUE") %>%
dplyr::select(-term)
})
mutate(bind_rows(result), pathway=names(result))## # A tibble: 14 x 5
## estimate std.error statistic p.value pathway
## <dbl> <dbl> <dbl> <dbl> <chr>
## 1 8.89 0.830 10.7 0.0000392 Androgen
## 2 1.25 3.22 0.389 0.711 EGFR
## 3 -6.90 0.830 -8.31 0.000165 Estrogen
## 4 0.783 1.92 0.408 0.698 Hypoxia
## 5 -0.972 0.657 -1.48 0.190 JAK-STAT
## 6 1.04 1.34 0.774 0.469 MAPK
## 7 0.456 0.697 0.655 0.537 NFkB
## 8 -4.16 0.774 -5.37 0.00171 p53
## 9 -0.635 1.89 -0.337 0.748 PI3K
## 10 2.34 1.09 2.15 0.0756 TGFb
## 11 0.715 0.681 1.05 0.335 TNFa
## 12 0.257 0.792 0.324 0.757 Trail
## 13 0.703 0.705 0.996 0.358 VEGF
## 14 -1.75 0.566 -3.10 0.0211 WNTWhat we see is that indeed the p53/DNA damage response pathway is less active after treatment than before.
Reproducing drug associations on the GDSC panel
Below is an example on how to calculate pathway scores for cell lines in the Genomics of Drug Sensitivity in Cancer (GDSC) panel, and to check for associations with drug response.
The code used for the analyses is available on Github.
Getting the data
This example shows how to use the GDSC gene expression data of multiple cell lines together with PROGENy to calculate pathway activity and then to check for associations with drug sensitivity.
First, we need the GDSC data for both gene expression and drug response. They are available on the GDSC1000 web site:
# set up a file cache so we download only once
library(BiocFileCache)
bfc = BiocFileCache(".")
# gene expression and drug response
base = "http://www.cancerrxgene.org/gdsc1000/GDSC1000_WebResources/Data/"
paths = bfcrpath(bfc, paste0(base, c("suppData/TableS4A.xlsx",
"preprocessed/Cell_line_RMA_proc_basalExp.txt.zip")))You can also download the files manually (adjust the file names when loading):
Creating the right objects to work with
First, we need to load the files we just downloaded into R to be able to perform the analysis:
# load the downloaded files
drug_table <- readxl::read_excel(paths[1], skip=5, na="NA")
drug_table <- replace(drug_table, is.na(drug_table), 0)
gene_table <- readr::read_tsv(paths[2])
# we need drug response with COSMIC IDs
drug_response <- data.matrix(drug_table[,3:ncol(drug_table)])
rownames(drug_response) <- drug_table[[1]]
# we need genes in rows and samples in columns
gene_expr <- data.matrix(gene_table[,3:ncol(gene_table)])
colnames(gene_expr) <- sub("DATA.", "", colnames(gene_expr), fixed=TRUE)
rownames(gene_expr) <- gene_table$GENE_SYMBOLSRunning PROGENy to get pathway activity scores
Activity inference is done using a weighted sum of the model genes. We can run this without worrying about the order of genes in the expression matrix using:
library(progeny)
pathways <- progeny(gene_expr,scale = TRUE, organism = "Human", top = 100,
perm = 1, verbose = FALSE)To visualize the progeny result
library(pheatmap)
myColor = colorRampPalette(c("Darkblue", "white","red"))(100)
pheatmap(pathways,fontsize=14, show_rownames = FALSE,
color=myColor, main = "PROGENy", angle_col = 45, treeheight_col = 0,
border_color = NA)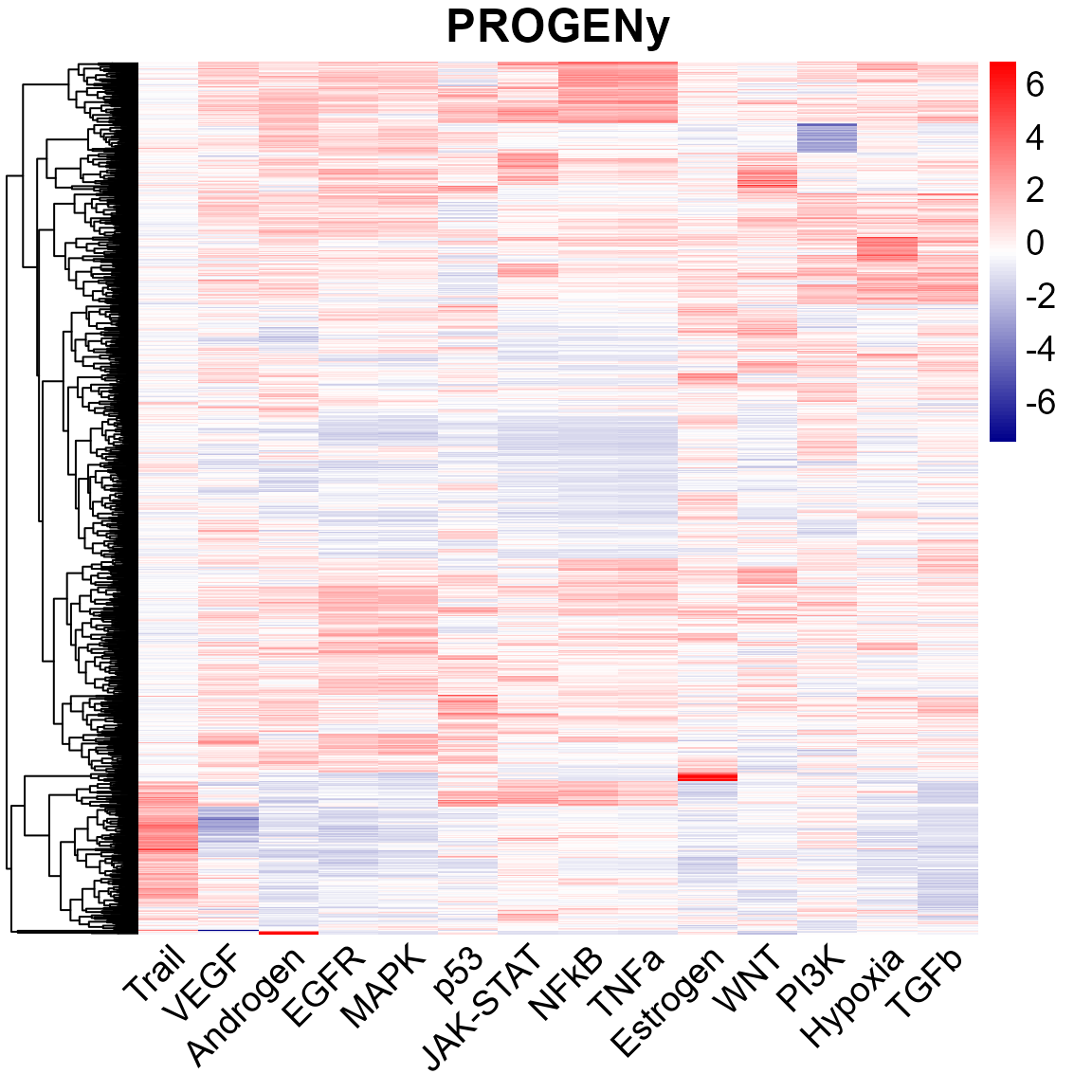
We now have the pathway activity scores for the pathways defined in PROGENy:
head(pathways)## Androgen EGFR Estrogen Hypoxia JAK-STAT MAPK
## 906826 -0.4241254 0.1385179 -0.269674710 -0.06875973 -0.37598907 0.41134699
## 687983 -1.7393225 -0.9213671 -0.763490640 -1.32374232 -0.94662180 -0.90704066
## 910927 -1.5393075 -0.0695914 1.613512221 -0.76482836 -1.05428545 0.17412564
## 1240138 0.9107488 -0.2200467 0.219584368 -0.73615734 -0.07536995 -0.60238190
## 1240139 -1.0602951 -0.1846655 -0.556250681 0.13664228 -0.58644833 -0.04550095
## 906792 0.7518034 0.8045299 0.008914293 0.40118664 -0.50038060 0.77199885
## NFkB p53 PI3K TGFb TNFa Trail
## 906826 -0.6374683 -1.0903096 -0.1953806 -0.7793323 -0.4329185 -0.8018626
## 687983 -1.3157127 -0.8692247 0.2542425 -0.8242281 -1.0195560 -0.5104074
## 910927 -0.3059911 0.8896616 -0.7487845 0.7580845 -0.1756034 -0.8092882
## 1240138 0.9755970 2.0632167 0.4839630 1.9108565 0.9865983 -0.2377919
## 1240139 -0.4751547 0.8763231 1.3814461 0.9999634 -0.3916422 -0.9224493
## 906792 -0.1878324 -0.8895494 -3.0379834 -0.3389734 -0.1436331 0.2455631
## VEGF WNT
## 906826 -0.13639016 0.01831199
## 687983 -0.08663407 -0.72986989
## 910927 0.13321227 2.28602257
## 1240138 0.01391272 1.51533198
## 1240139 0.90296847 1.71036886
## 906792 0.18149340 -1.22160214Testing if MAPK activity is significantly associated with Trametinib
Trametinib is a MEK inhibitor, so we would assume that cell lines that have a higher MAPK activity are more sensitive to MEK inhibition.
We can test this the following way:
cell_lines = intersect(rownames(pathways), rownames(drug_response))
trametinib = drug_response[cell_lines, "Trametinib"]
mapk = pathways[cell_lines, "MAPK"]
associations = lm(trametinib ~ mapk)
summary(associations)##
## Call:
## lm(formula = trametinib ~ mapk)
##
## Residuals:
## Min 1Q Median 3Q Max
## -6.548 -1.255 0.338 1.338 6.819
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -0.94703 0.06442 -14.70 <2e-16 ***
## mapk -1.35978 0.06449 -21.09 <2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 1.998 on 960 degrees of freedom
## Multiple R-squared: 0.3165, Adjusted R-squared: 0.3158
## F-statistic: 444.6 on 1 and 960 DF, p-value: < 2.2e-16And indeed we find that MAPK activity is strongly associated with sensitivity to Trametinib: the Pr(>|t|) is much smaller than the conventional threshold of 0.05.
The intercept is significant as well, but we're not really interested if the mean drug response is above or below 0 in this case.
Note, however, that we tested all cell lines at once and did not adjust for the effect different tissues may have.
R version information
## R version 4.0.4 (2021-02-15)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 10 x64 (build 18363)
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=English_Germany.1252 LC_CTYPE=English_Germany.1252
## [3] LC_MONETARY=English_Germany.1252 LC_NUMERIC=C
## [5] LC_TIME=English_Germany.1252
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] pheatmap_1.0.12 BiocFileCache_1.14.0
## [3] dbplyr_2.1.1 dplyr_1.0.6
## [5] progeny_1.15.1 biomaRt_2.46.3
## [7] DESeq2_1.30.1 airway_1.10.0
## [9] SummarizedExperiment_1.20.0 Biobase_2.50.0
## [11] GenomicRanges_1.42.0 GenomeInfoDb_1.26.7
## [13] IRanges_2.24.1 S4Vectors_0.28.1
## [15] BiocGenerics_0.36.1 MatrixGenerics_1.2.1
## [17] matrixStats_0.59.0 knitr_1.33
## [19] BiocStyle_2.18.1
##
## loaded via a namespace (and not attached):
## [1] colorspace_2.0-1 ellipsis_0.3.2 rprojroot_2.0.2
## [4] XVector_0.30.0 fs_1.5.0 rstudioapi_0.13
## [7] ggrepel_0.9.1 bit64_4.0.5 AnnotationDbi_1.52.0
## [10] fansi_0.5.0 xml2_1.3.2 splines_4.0.4
## [13] cachem_1.0.5 geneplotter_1.68.0 jsonlite_1.7.2
## [16] broom_0.7.7 annotate_1.68.0 readr_1.4.0
## [19] BiocManager_1.30.16 compiler_4.0.4 httr_1.4.2
## [22] backports_1.2.1 assertthat_0.2.1 Matrix_1.3-4
## [25] fastmap_1.1.0 cli_2.5.0 htmltools_0.5.1.1
## [28] prettyunits_1.1.1 tools_4.0.4 gtable_0.3.0
## [31] glue_1.4.2 GenomeInfoDbData_1.2.4 rappdirs_0.3.3
## [34] Rcpp_1.0.6 cellranger_1.1.0 jquerylib_0.1.4
## [37] pkgdown_1.6.1 vctrs_0.3.8 xfun_0.24
## [40] stringr_1.4.0 ps_1.6.0 lifecycle_1.0.0
## [43] XML_3.99-0.6 zlibbioc_1.36.0 scales_1.1.1
## [46] ragg_1.1.3 hms_1.1.0 RColorBrewer_1.1-2
## [49] yaml_2.2.1 curl_4.3.1 memoise_2.0.0
## [52] gridExtra_2.3 ggplot2_3.3.4 sass_0.4.0
## [55] stringi_1.6.2 RSQLite_2.2.7 highr_0.9
## [58] genefilter_1.72.1 desc_1.3.0 BiocParallel_1.24.1
## [61] rlang_0.4.11 pkgconfig_2.0.3 systemfonts_1.0.2
## [64] bitops_1.0-7 evaluate_0.14 lattice_0.20-44
## [67] purrr_0.3.4 bit_4.0.4 tidyselect_1.1.1
## [70] magrittr_2.0.1 bookdown_0.22 R6_2.5.0
## [73] generics_0.1.0 DelayedArray_0.16.3 DBI_1.1.1
## [76] pillar_1.6.1 withr_2.4.2 survival_3.2-11
## [79] RCurl_1.98-1.3 tibble_3.1.2 crayon_1.4.1
## [82] utf8_1.2.1 rmarkdown_2.9 progress_1.2.2
## [85] locfit_1.5-9.4 grid_4.0.4 readxl_1.3.1
## [88] blob_1.2.1 digest_0.6.27 xtable_1.8-4
## [91] tidyr_1.1.3 textshaping_0.3.5 openssl_1.4.4
## [94] munsell_0.5.0 bslib_0.2.5.1 askpass_1.1