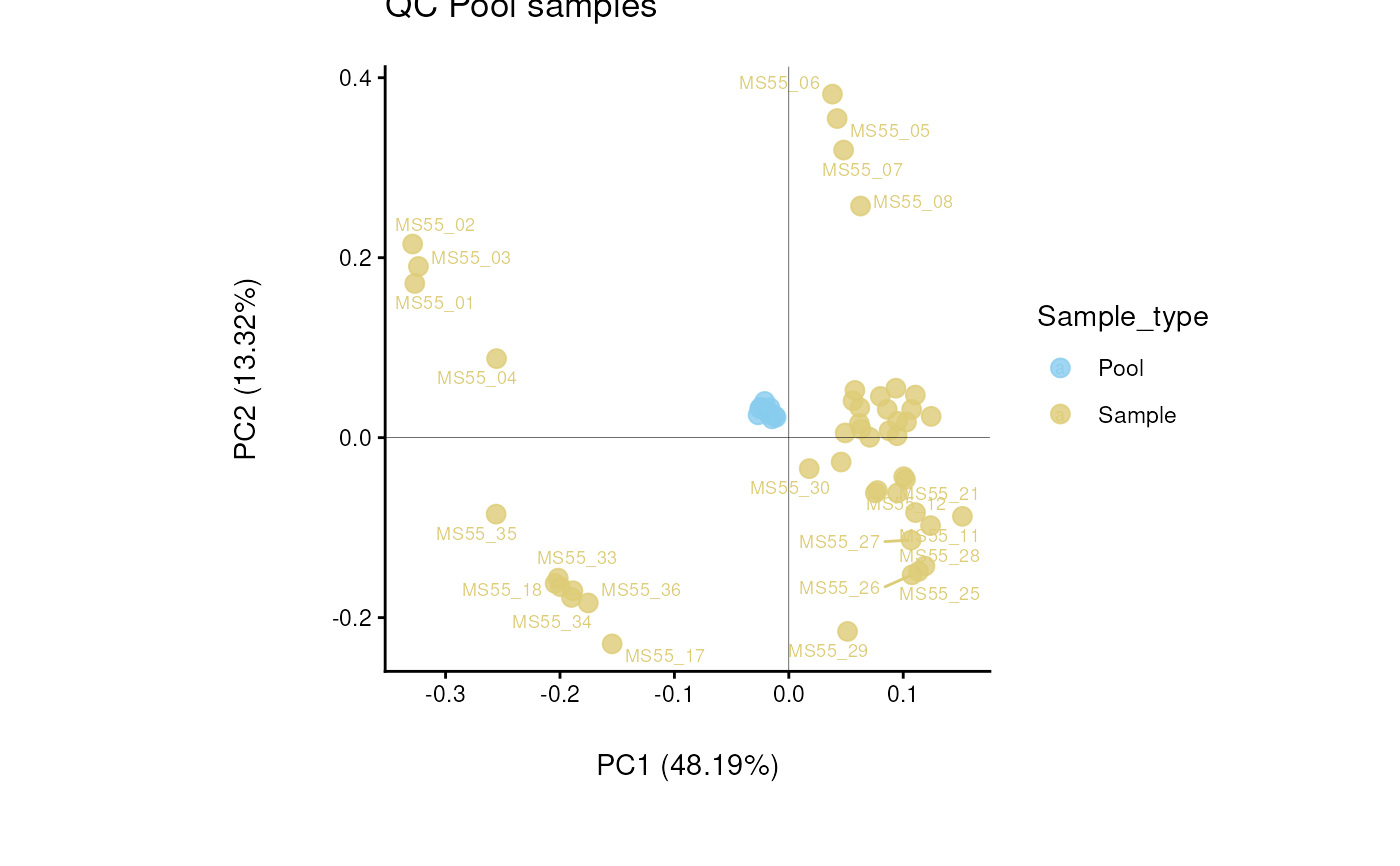
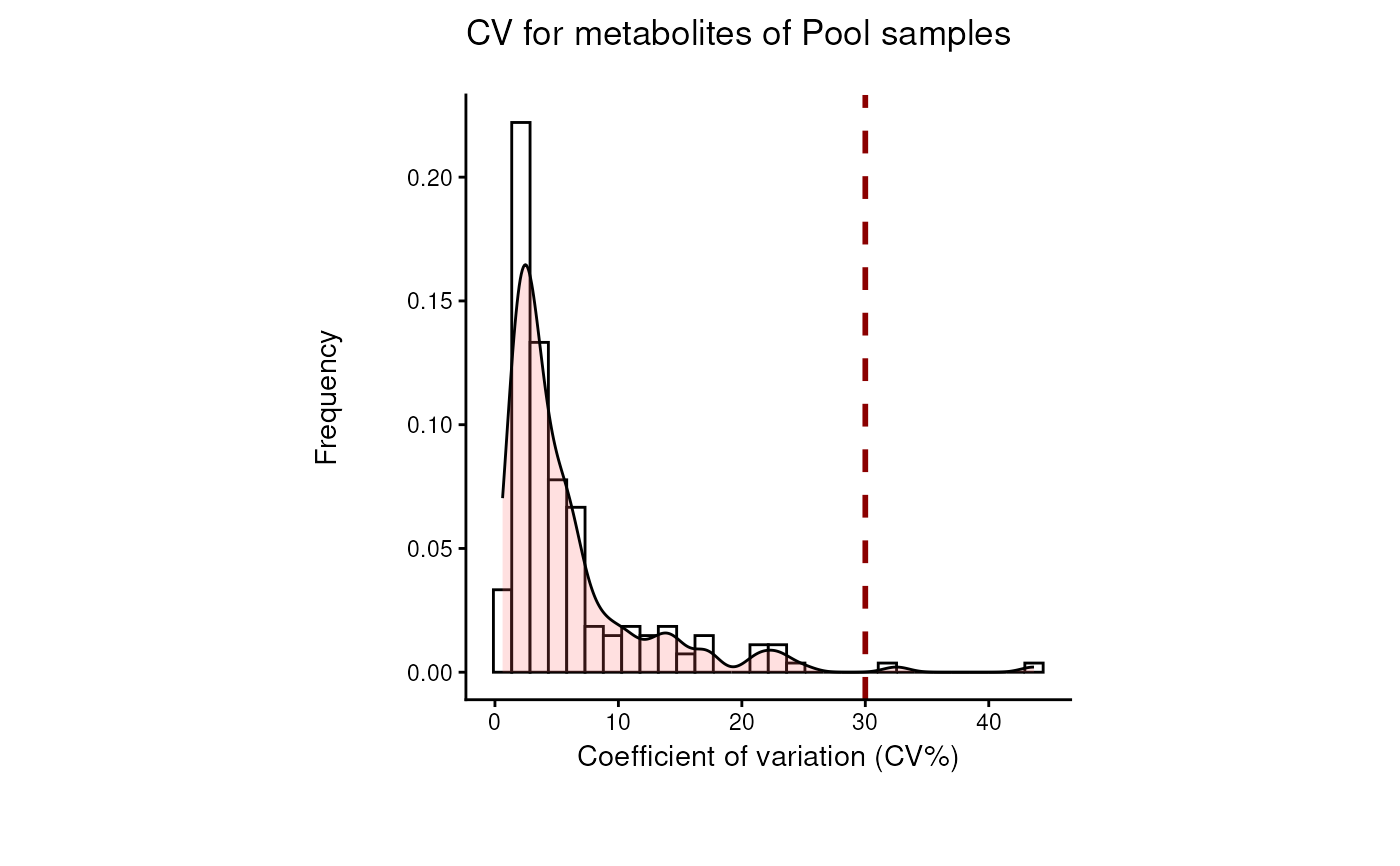
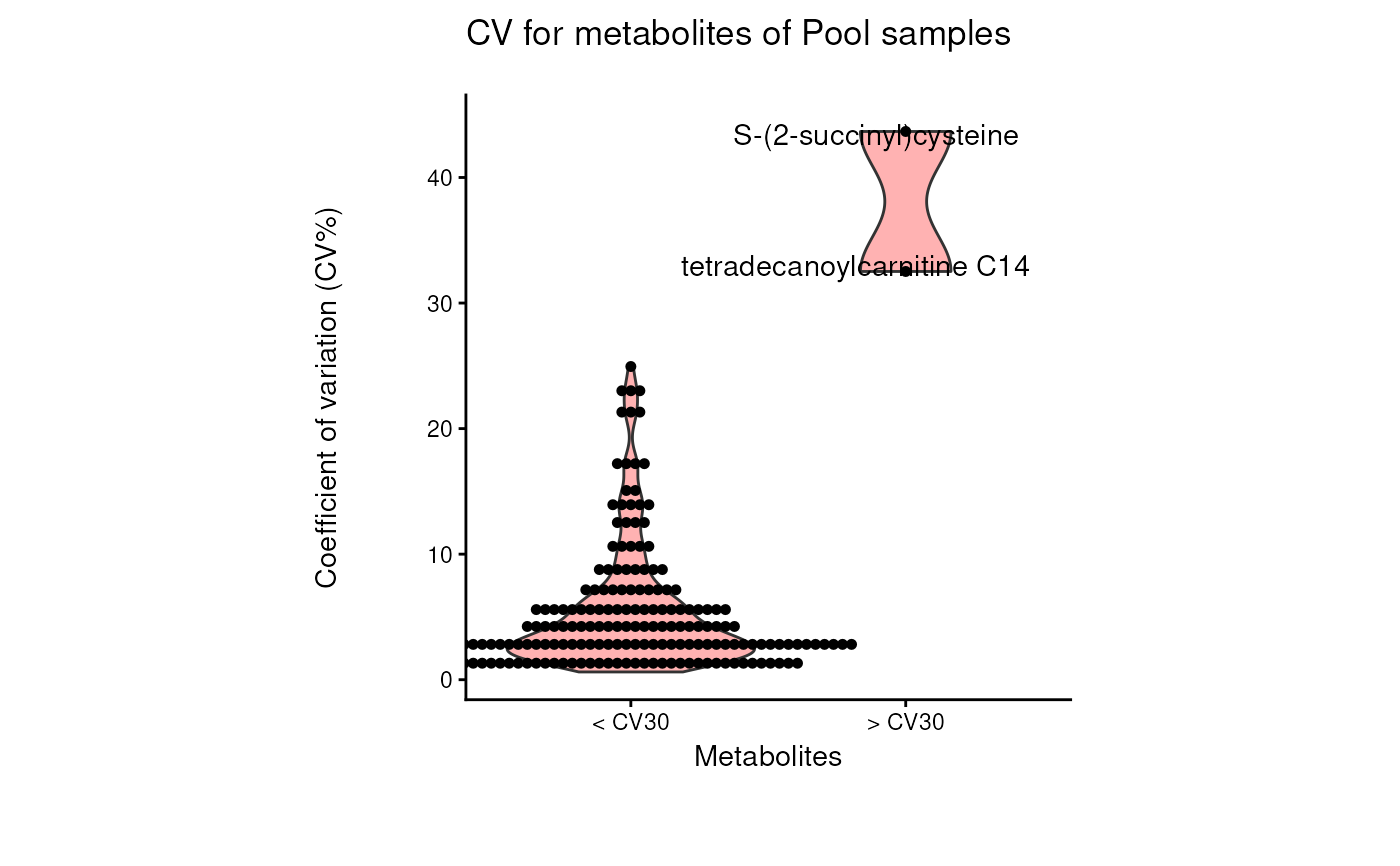
Find metabolites with high variability across total pool samples
Source:R/Processing.R
pool_estimation.RdFind metabolites with high variability across total pool samples
Usage
pool_estimation(
data,
metadata_sample = NULL,
metadata_info = NULL,
cutoff_cv = 30,
save_plot = "svg",
save_table = "csv",
print_plot = TRUE,
path = NULL
)Arguments
- data
SummarizedExperiment (se) file including assay and colData. If se file is provided, metadata_sample is extracted from the colData of the se object. Alternatively provide a DF which contains unique sample identifiers as row names and metabolite numerical values in columns with metabolite identifiers as column names. Use NA for metabolites that were not detected.
- metadata_sample
Optional: Only required if you did not provide se file in parameter data. Provide DF which contains information about the samples, which will be combined with the input data based on the unique sample identifiers used as rownames. Must contain column with Conditions. If you do not have multiple conditions in your experiment assign all samples into the same condition. Default = NULL
- metadata_info
Optional: NULL or Named vector including the Conditions and PoolSample information (Name of the Conditions column and Name of the pooled samples in the Conditions in the Input_SettingsFile) : c(Conditions="ColumnNameConditions, PoolSamples=NamePoolCondition. If no Conditions is added in the Input_metadata_info, it is assumed that the conditions column is named 'Conditions' in the Input_SettingsFile. ). Default = NULL
- cutoff_cv
Optional: Filtering cutoff for high variance metabolites using the Coefficient of Variation. Default = 30
- save_plot
Optional: Select the file type of output plots. Options are svg, png, pdf or NULL. Default = svg
- save_table
Optional: File types for the analysis results are: "csv", "xlsx", "txt", ot NULL default: "csv"
- print_plot
Optional: If TRUE prints an overview of resulting plots. Default = TRUE
- path
Optional: Path to the folder the results should be saved at. default: NULL
Value
List with two elements: DF (including input and output table) and Plot (including all plots generated)
Examples
data(intracell_raw_se)
Res <- pool_estimation(
data = intracell_raw_se,
metadata_info = c(PoolSamples = "Pool", Conditions = "Conditions")
)
#> Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
#> ℹ Please use tidy evaluation idioms with `aes()`.
#> ℹ See also `vignette("ggplot2-in-packages")` for more information.
#> ℹ The deprecated feature was likely used in the ggfortify package.
#> Please report the issue at <https://github.com/sinhrks/ggfortify/issues>.
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?
#> Bin width defaults to 1/30 of the range of the data. Pick better value with
#> `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?
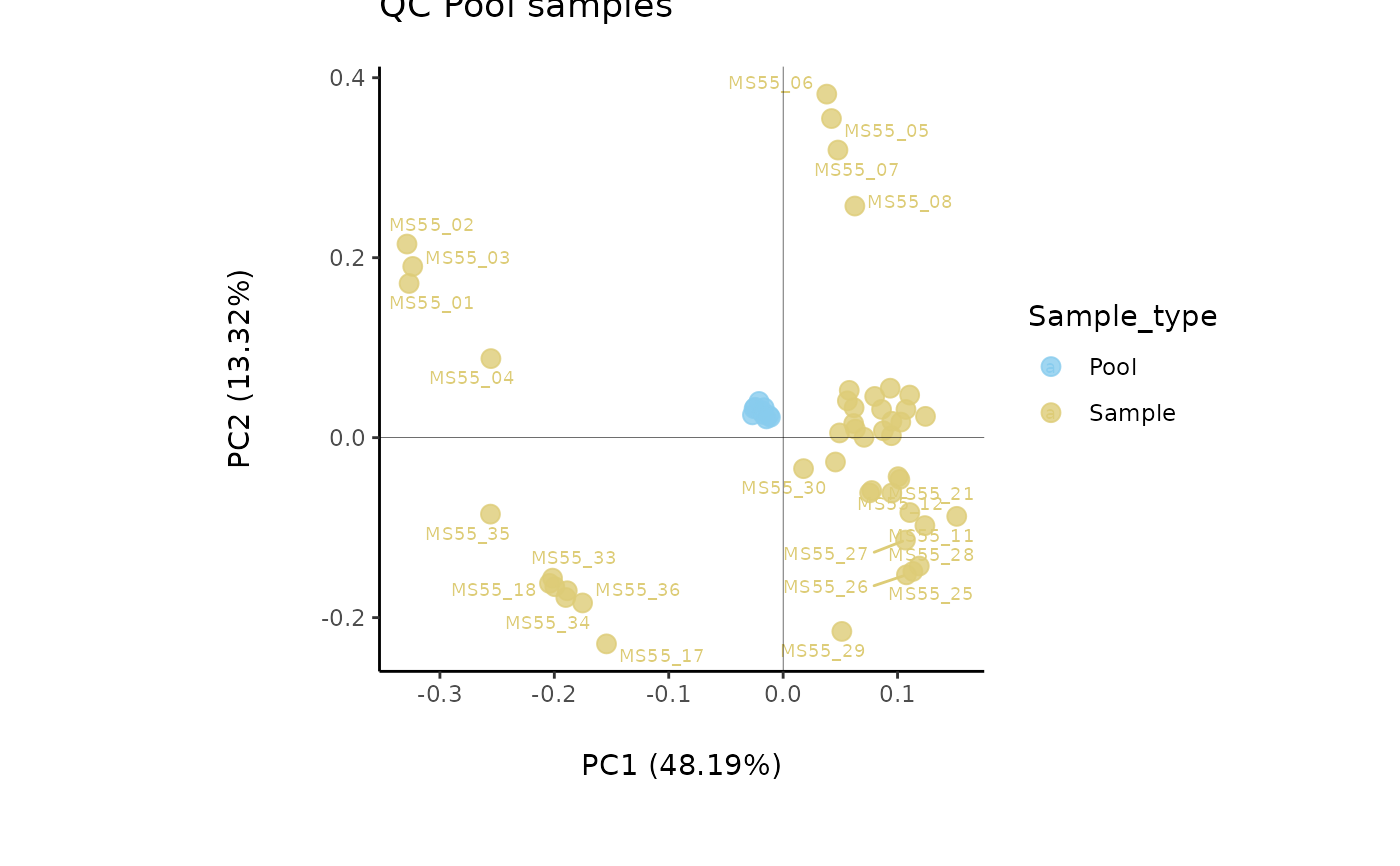
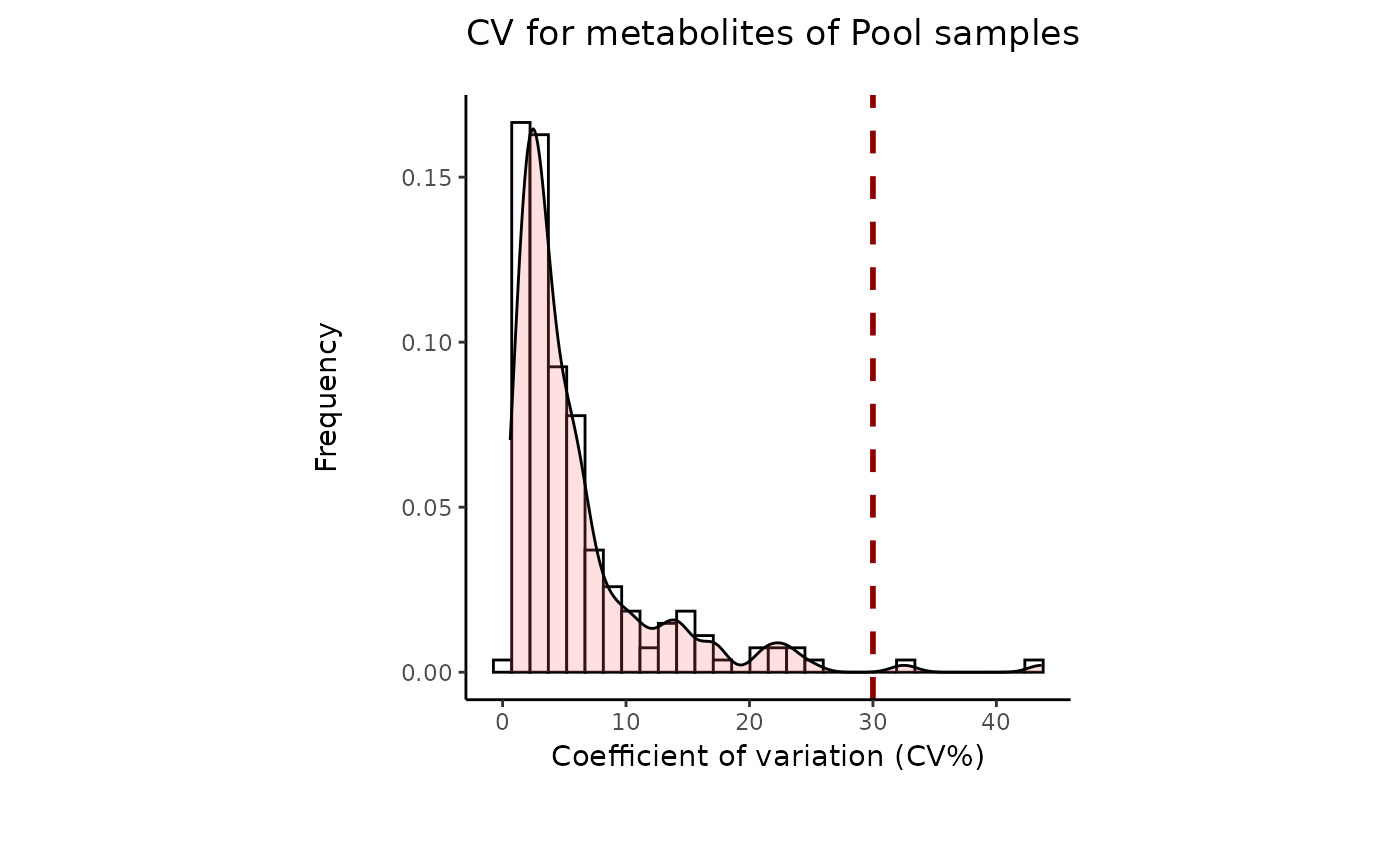
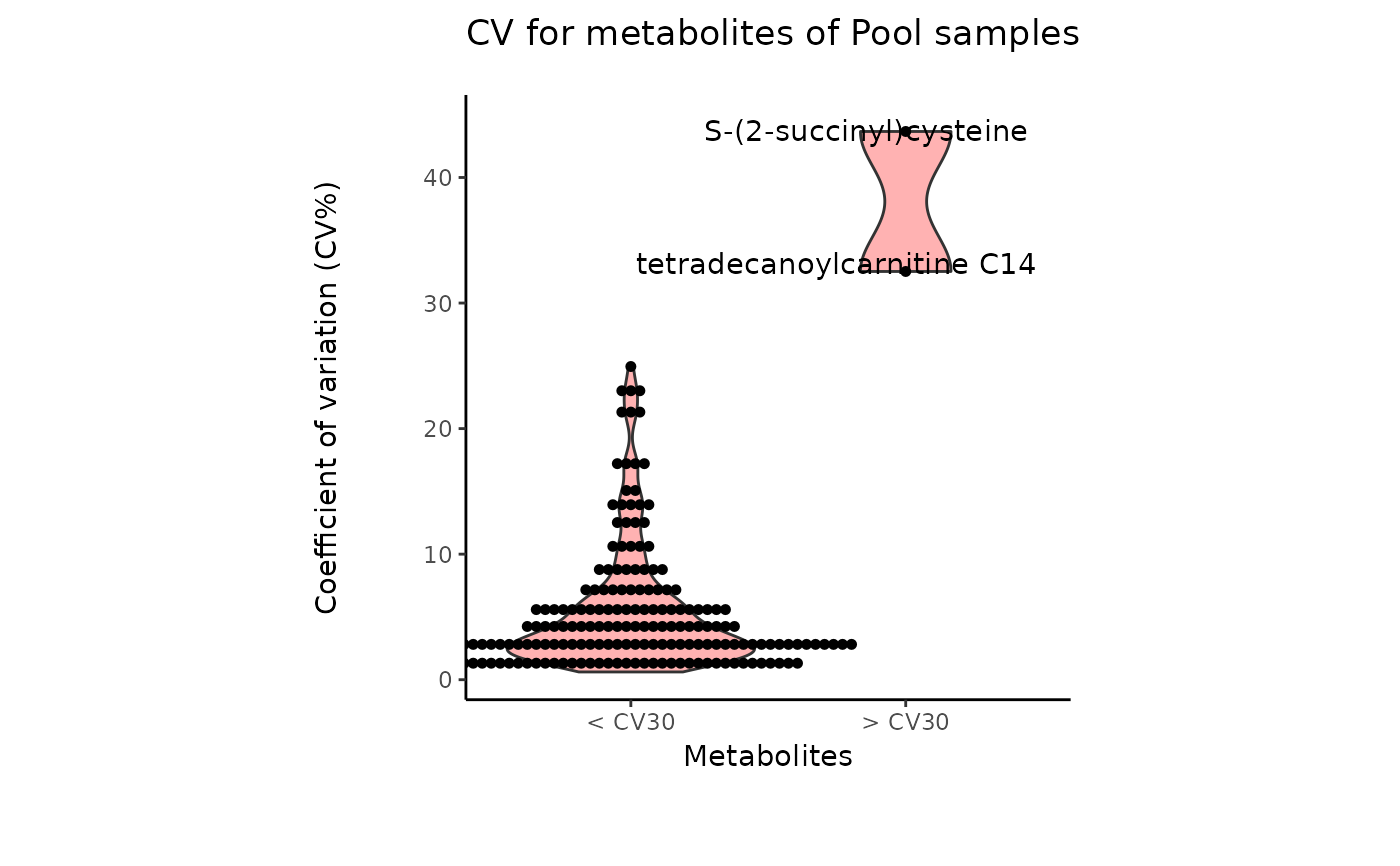 data(intracell_raw)
Intra <- intracell_raw %>% tibble::column_to_rownames("Code")
Res <- pool_estimation(
data = Intra[, -c(1:3)],
metadata_sample = Intra[, c(1:3)],
metadata_info = c(PoolSamples = "Pool", Conditions = "Conditions")
)
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?
#> Bin width defaults to 1/30 of the range of the data. Pick better value with
#> `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?
data(intracell_raw)
Intra <- intracell_raw %>% tibble::column_to_rownames("Code")
Res <- pool_estimation(
data = Intra[, -c(1:3)],
metadata_sample = Intra[, c(1:3)],
metadata_info = c(PoolSamples = "Pool", Conditions = "Conditions")
)
#> `stat_bin()` using `bins = 30`. Pick better value `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?
#> Bin width defaults to 1/30 of the range of the data. Pick better value with
#> `binwidth`.
#> Warning: The following aesthetics were dropped during statistical transformation: label.
#> ℹ This can happen when ggplot fails to infer the correct grouping structure in
#> the data.
#> ℹ Did you forget to specify a `group` aesthetic or to convert a numerical
#> variable into a factor?