COSMOS-tutorial
A. Dugourd, A. Gabor and K. Zirngibl
11/10/2020
Source:vignettes/tutorial.Rmd
tutorial.RmdInstallation and dependency
COSMOS is dependent on CARNIVAL for exhibiting the signalling pathway optimisation. CARNIVAL requires the interactive version of IBM Cplex or CBC-COIN solver as the network optimiser. The IBM ILOG Cplex is freely available through Academic Initiative here. The CBC solver is open source and freely available for any user, but has a significantly lower performance than CPLEX. Obtain CBC executable directly usable for cosmos here. Alternatively for small networks, users can rely on the freely available lpSolve R-package, which is automatically installed with the package.
In this tutorial we use lpSolve, but we strongly recommend to obtain a license for CPLEX.
# install from bioconductor
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("cosmosR")
# install the newest (development) version from GitHub
# install.packages("remotes")
# install CARNIVAL from github
remotes::install_github("saezlab/CARNIVAL")
remotes::install_github("saezlab/cosmosR")Introduction
COSMOS (Causal Oriented Search of Multi-Omic Space) is a method that integrates phosphoproteomics, transcriptomics, and metabolomics data sets. COSMOS leverages extensive prior knowledge of signaling pathways, metabolic networks, and gene regulation with computational methods to estimate activities of transcription factors and kinases as well as network-level causal reasoning. This pipeline can provide mechanistic explanations for experimental observations across multiple omic data sets.
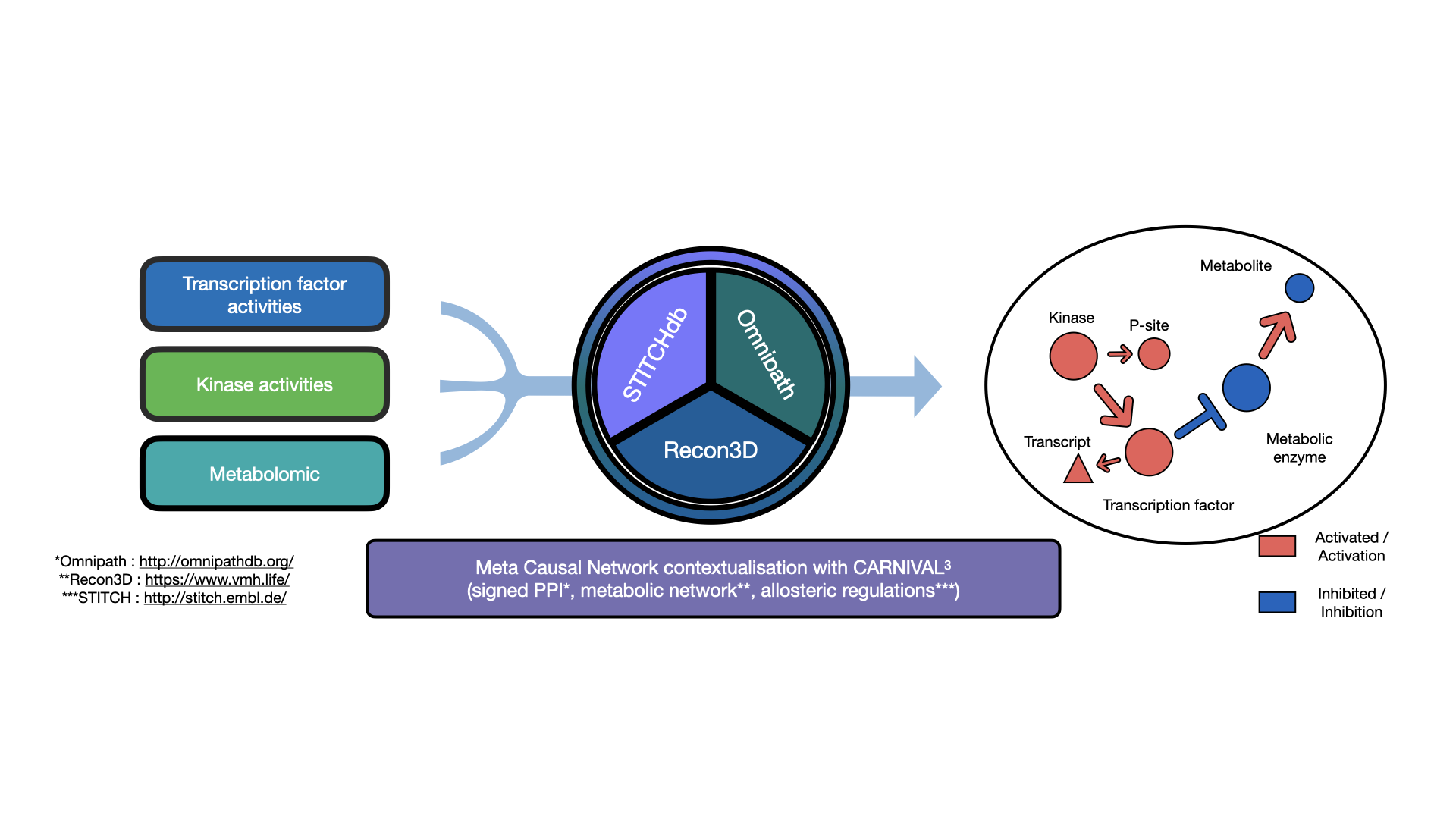
Essentially, COSMOS has 3 main components:
The prior knowledge: The goal of COSMOS is essentially to connect various deregulation events together with mechanistic hypotheses. Those mechanistic hypotheses are basically known potential interactions between molecular biology features, such as proteins and metabolites. An example of such interaction can be the activation of MTOR by AKT, in the EGFR canonical pathway. Thousands of such potential interaction can be assembled together to form a large network of interactions. Such networks are thus called prior knowledge networks (PKN), because they summarize large amounts of prior knowledge in the form of a network. In the context of COSMOS this interactions come from the Omnipath database, STICHdb and the recon3D reaction network. With respect to the example interaction between MTOR and AKT, the question that is usually asked when presented with a given experimental context is: while MTOR can activate AKT in general, is this interaction relevant in my experimental context? This brings us to the second component of COSMOS.
The data: Information about deregulated transcription factors (TF), kinases/phosphophatases and metabolites can be extracted from transcriptomic, phospho-proteomic or metabolomic data respectively, using footprint analysis methods (see:https://github.com/saezlab/transcriptutorial (Especially chapter 4)). Such information can be quite useful by itself, to characterize which TFs or kinases are specifically deregulated in cancer cells for example. the goal of COSMOS is to find how these different deregulation events can be connected together. Going back to the example of MTOR and AKT, if I know that AKT can potentially activate MTOR, and I observe in my data that both AKT and MTOR are significantly activated in cancer tissue compared to their healthy counterpart, there is a reasonable chance that this interaction is active in the cancer tissue I’m studying. The problem is that data often contain information about hundreds of up and down-regulated activities at the same time, and thousands of potential interactions can potentially explain them. In order to find the optimal set of interactions that are relevant to explain the deregulation observed in my data, an algorithm is needed.
The algorithm: The problem described in the data part is formulated as an integer linear optimisation problem. The PKN and the data can be abstracted as a large set of integer variables operating together in an even larger set of equations (see https://www.nature.com/articles/s41540-019-0118-z for more info). Given a set of fixed value for the variable (that is, a subset of the known deregulation), we need to find the values for the other sets of variable (corresponding to proteins and metabolites for which we have no information in our data) that can lead to a satisfying result. A satisfying results in the context of COSMOS represent a sub-network of interactions that is as small as possible while coherently explaining as many of our known deregulation as possible. Thus, the set of equation mentioned earlier needs to be solved, using an integer linear programming solver. The solvers that are currently available for COSMOS are lpsolve, cbc and cplex. In general, CPLEX is preferred over CBC for most real case applications, while lpsolve is mainly used for testing with small networks and sets of measurements.
Tutorial section: signaling to metabolism
First, we load the package
In this part, we can set up the options for the CARNIVAL run, such as timelimit and min gap tolerance.
The user should provide a path to its CPLEX/cbc executable.
You can check the CARNIVAL_options variable to see all possible options that can be adjusted
In this example, we will use the built-in solver lpSolve. User should be aware that lpSolve should ONLY be used for TESTS. To obtain meaningful results, best solver is cplex, or cbc if not possible.
CARNIVAL_options <- default_CARNIVAL_options(solver = "lpSolve")
# To use CBC
# CARNIVAL_options <- default_CARNIVAL_options(solver = "cbc")
# CARNIVAL_options$solverPath <- "~/Documents/cbc"
# CARNIVAL_options$threads <- 2
# CARNIVAL_options$mipGAP <- 0.05
# To use CPLEX:
# CARNIVAL_options <- default_CARNIVAL_options(solver = "cplex")
# CARNIVAL_options$solverPath <- "C:/Program Files/CPLEX_solver/cplex/bin/x64_win64/cplex.exe"
# CARNIVAL_options$threads <- 2
# CARNIVAL_options$mipGAP <- 0.05
CARNIVAL_options$timelimit <- 3600In the next section, we prepare the input to run cosmosR. The signaling inputs are the result of footprint based TF and kinase activity estimation. For more info on TF activity estimation from transcriptomic data, see:https://github.com/saezlab/transcriptutorial (Especially chapter 4)
Here we use of toy PKN, to see the full meta PKN, you can load it with data(meta_network). To see how is the meta_PKN assembled, see: https://github.com/saezlab/meta_PKN_BIGG.git
The metabolites in the prior knowledge network are identified as XMetab__HMDBid_compartment or XMetab__BIGGid_compartment (for example “Metab__HMDB0000190_c”). The compartment code is the BIGG model standard (r, c, e, x, m, l, n, g). Thus we will first need to map whatever identifier for metabolite the data has to the one of the network. Genes are identified as gene symboles (in the signaling part of network) or Gene####__symbole (in the reaction network part of network).
The maximum network depth will define the maximum number of step downstream of kinase/TF COSMOS will look for deregulated metabolites. Good first guess for max depth could be around between 4 and 6 (here it is 15 for the toy dataset)
The differential experession data is used to filter out wrong TF-target interactions in this context after a pre-optimisation.
The list of genes in the differential expression data will also be used as a reference to define which genes are expressed or not (all genes in the diff_expression_data are considered expressed, and genes that are no in diff_expression_data are removed from the network).
Here, the CARNIVAL_options$timelimit is set for the pre-optimisation. Indeed, if the “filter_tf_gene_interaction_by_optimization” parameter of the “preprocess_COSMOS_signaling_to_metabolism” function is set to TRUE, COSMOS can perform a first optimisation run in order to generate a preliminary solution network that can be used for filtering out incoherences between TF activities and downstream target expressions.
data(toy_network)
data(toy_signaling_input)
data(toy_metabolic_input)
data(toy_RNA)
test_for <- preprocess_COSMOS_signaling_to_metabolism(meta_network = toy_network,
signaling_data = toy_signaling_input,
metabolic_data = toy_metabolic_input,
diff_expression_data = toy_RNA,
maximum_network_depth = 15,
remove_unexpressed_nodes = TRUE,
CARNIVAL_options = CARNIVAL_options)## [1] "COSMOS: all 3 signaling nodes from data were found in the meta PKN"
## [1] "COSMOS: all 2 metabolic nodes from data were found in the meta PKN"
## [1] "COSMOS: 2975 of the 9300 genes in expression data were found as transcription factor target"
## [1] "COSMOS: 2975 of the 5321 transcription factor targets were found in expression data"
## [1] "COSMOS: removing unexpressed nodes from PKN..."
## [1] "COSMOS: 0 interactions removed"
## [1] "COSMOS: removing nodes that are not reachable from inputs within 15 steps"
## [1] "COSMOS: 0 from 101 interactions are removed from the PKN"
## [1] "COSMOS: removing nodes that are not observable by measurements within 15 steps"
## [1] "COSMOS: 52 from 101 interactions are removed from the PKN"
## [1] "COSMOS: 2 input/measured nodes are not in PKN any more: USF1, SRF and 0 more."
## [1] "COSMOS: 0 interactions are removed from the PKN based on consistency check between TF activity and gene expression"
## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."
## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."## [1] "COSMOS: 0 interactions are removed from the PKN based on consistency check between TF activity and gene expression"
## [1] "COSMOS: all 1 signaling nodes from data were found in the meta PKN"
## [1] "COSMOS: all 2 metabolic nodes from data were found in the meta PKN"
## [1] "COSMOS: 2975 of the 9300 genes in expression data were found as transcription factor target"
## [1] "COSMOS: 2975 of the 5321 transcription factor targets were found in expression data"In this part, we can set up the options for the actual run, such as timelimit and min gap tolerance.
The running time should be much higher here than in pre-optimisation. You can increase the number of threads to use if you have many available CPUs.
CARNIVAL_options$timelimit <- 14400
CARNIVAL_options$mipGAP <- 0.05
CARNIVAL_options$threads <- 2This is where cosmosR run.
test_result_for <- run_COSMOS_signaling_to_metabolism(data = test_for,
CARNIVAL_options = CARNIVAL_options)## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."Finally, we process the results of the first cosmosR run, to translate gene names and metabolites name.
formated_result_for <- format_COSMOS_res(test_result_for)Tutorial section: metabolism to signaling
Before we run the metabolism to signaling part, we need to prepare again the inputs.
CARNIVAL_options$timelimit <- 3600
CARNIVAL_options$mipGAP <- 0.05
CARNIVAL_options$threads <- 2Now that the correct time is set up for the pre-optimisation run, we can prepare the inputs.
test_back <- preprocess_COSMOS_metabolism_to_signaling(meta_network = toy_network,
signaling_data = toy_signaling_input,
metabolic_data = toy_metabolic_input,
diff_expression_data = toy_RNA,
maximum_network_depth = 15,
remove_unexpressed_nodes = FALSE,
CARNIVAL_options = CARNIVAL_options)## [1] "COSMOS: all 3 signaling nodes from data were found in the meta PKN"
## [1] "COSMOS: all 2 metabolic nodes from data were found in the meta PKN"
## [1] "COSMOS: 2975 of the 9300 genes in expression data were found as transcription factor target"
## [1] "COSMOS: 2975 of the 5321 transcription factor targets were found in expression data"
## [1] "COSMOS: removing nodes that are not reachable from inputs within 15 steps"
## [1] "COSMOS: 0 from 101 interactions are removed from the PKN"
## [1] "COSMOS: removing nodes that are not observable by measurements within 15 steps"
## [1] "COSMOS: 54 from 101 interactions are removed from the PKN"
## [1] "COSMOS: 1 input/measured nodes are not in PKN any more: Metab__HMDB0000190_c and 0 more."
## [1] "COSMOS: 0 interactions are removed from the PKN based on consistency check between TF activity and gene expression"
## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."
## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."## [1] "COSMOS: 0 interactions are removed from the PKN based on consistency check between TF activity and gene expression"
## [1] "COSMOS: all 3 signaling nodes from data were found in the meta PKN"
## [1] "COSMOS: all 1 metabolic nodes from data were found in the meta PKN"
## [1] "COSMOS: 2975 of the 9300 genes in expression data were found as transcription factor target"
## [1] "COSMOS: 2975 of the 5321 transcription factor targets were found in expression data"Then we can run cosmosR to connect metabolism to signaling. The running time here usually needs to be longer, as this problem seems to be harder to solve for CPLEX.
CARNIVAL_options$timelimit <- 28800
test_result_back <- run_COSMOS_metabolism_to_signaling(data = test_back,
CARNIVAL_options = CARNIVAL_options)## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."## [1] "lpSolve does not scale well with large PKNs. This solver is mainly for testing purposes. To run COSMSO, we recommend using cplex, or cbc solvers."Finally we can format the result of the backward run as well (same as for forward run)
formated_result_back <- format_COSMOS_res(test_result_back)Tutorial section: Merge forward and backward networks and visualise network
Here we simply take the union of forward and backward runs to create a full network solution lopping between signaling, gene-regulation and metabolism. Since there is an overlap between the result network of forward and backward run, you may optionally want to check if there are any node sign that are incoherent in the overlap between the two solutions.
full_sif <- as.data.frame(rbind(formated_result_for[[1]], formated_result_back[[1]]))
full_sif <- full_sif[full_sif$Weight>0,]
full_attributes <- as.data.frame(rbind(formated_result_for[[2]], formated_result_back[[2]]))
full_sif <- unique(full_sif)
full_attributes <- unique(full_attributes)This function will generate a dynamic network plot centered on a given node of the network solution, and connecting it to measured nodes in the given range (here 7 steps).
network_plot <- display_node_neighboorhood(central_node = 'Metab__D-Glucitol_c',
sif = full_sif,
att = full_attributes,
n = 7)
network_plotHere is how this network can be intepreted (this is purely illustrative, as some of those interaction may be incorectly signed because lpsolve can only use positive interactions):
This network represents the flow of activities that can connect MYC up-regulation with Glucitol (Sorbitol) accumulation. Here, NFKB1 can upregulate the expression of SLC2A1, which in turn transport more glucose in the cytoplasm. The increase transport of glucose can lead to more glucose being avlaible for conversion into glucitol by the AKR1A enzyme. Interestingly, glucitol is a now activator of MAPK14, thus leading to the appearance of a positive feedback loop connecting MYC, glucitol and MAPK14.
It is important to understand that each of this links is hypothetical. The come from a larger pool of potential molecular interactions present in multiple online databases and compiled in omnipath, STITCH and recon metabolic network. They exist in the literature and are interactions that are known to potentially exists in other experimental contexts. Thus, COSMOS compile all those potential interactions together and proposes a coherent set that can explain the data at hand.
Those links should however be considered only as potential mechanistic connections, and will need to be further confirmed experimentally. Those interactions can be searched in the literature to see in which other disease or experimental context they have been shown to be relevant. Taken together, multiple interactions can help to build a biological story that can guide further underatanding of the underlying biology and decide on future experiments.
Tutorial section: Over Representation Analysis
Often it is useful to perform an Over Representation Analysis (ORA) on the resulting nodes of a COSMOS network as a first analysis step to get a more functional interpretation on the modeled signaling cascade. A common way to this is to test whether the selected genes (nodes) in the COSMOS solution network show statistically significant differences in comparison to the prior-knowledge network (PKN).
The differentially expressed genes give information about the cellular processes that are deregulated and if the proportions in various pathways are SIGNIFICANTLY different from what is expected.In this way the significant differences between two biological conditions (e.g. cancer vs. normal tissue, or treatment vs. untreated cells) can be shown.
Algorithms that perform an ORA are implemented in other R packages like piano or decoupleR. In addition to a gene set collection these algorithms require two different lists as inputs: - nodes in the COSMOS solution network which relate back to the input data (e.g. transcriptomics, proteomics, metabolomics, fluxomics, or perturbations) - all nodes (kinases, transcription factors, metabolites) in the prior-knowledge network (which are used as the background in our analysis)
In this section we will show how to obtain these two lists from a formated COSMOS result object.
sif_forward = formated_result_for[[1]]
att_forward = formated_result_for[[2]]
nodes_ORA = extract_nodes_for_ORA(
sif = sif_forward,
att = att_forward)Now this forward and backward sets can be used with any ORA analysis.
## R version 4.4.1 (2024-06-14)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 22.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so; LAPACK version 3.10.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] cosmosR_1.19.1
##
## loaded via a namespace (and not attached):
## [1] bcellViper_1.40.0 sass_0.4.10 generics_0.1.4
## [4] tidyr_1.3.2 lpSolve_5.6.23 stringi_1.8.7
## [7] lattice_0.22-9 hms_1.1.4 digest_0.6.39
## [10] magrittr_2.0.5 evaluate_1.0.5 grid_4.4.1
## [13] fastmap_1.2.0 jsonlite_2.0.0 Matrix_1.7-6
## [16] progress_1.2.3 purrr_1.2.2 codetools_0.2-20
## [19] textshaping_1.0.5 jquerylib_0.1.4 cli_3.6.6
## [22] rlang_1.3.0 decoupleR_2.10.0 crayon_1.5.3
## [25] visNetwork_2.1.4 parallelly_1.48.0 bit64_4.8.2
## [28] withr_3.0.3 cachem_1.1.0 yaml_2.3.12
## [31] otel_0.2.0 tools_4.4.1 parallel_4.4.1
## [34] tzdb_0.5.0 BiocParallel_1.38.0 dplyr_1.2.1
## [37] vctrs_0.7.3 R6_2.6.1 lifecycle_1.0.5
## [40] stringr_1.6.0 bit_4.6.0 fs_2.1.0
## [43] htmlwidgets_1.6.4 vroom_1.7.1 ragg_1.5.2
## [46] pkgconfig_2.0.3 desc_1.4.3 pkgdown_2.2.1
## [49] pillar_1.11.1 bslib_0.11.0 glue_1.8.1
## [52] systemfonts_1.3.2 xfun_0.60 tibble_3.3.1
## [55] tidyselect_1.2.1 knitr_1.51 CARNIVAL_2.14.0
## [58] dorothea_1.16.0 rjson_0.2.23 htmltools_0.5.9
## [61] igraph_2.3.3 rmarkdown_2.31 readr_2.2.0
## [64] compiler_4.4.1 prettyunits_1.2.0