Metadata Analysis
Christina Schmidt
Heidelberg UniversitySource:
vignettes/Metadata Analysis.Rmd
Metadata Analysis.Rmd
Tissue metabolomics experiment is a standard metabolomics experiment
using tissue samples (e.g. from animals or patients).
In this tutorial we showcase how
to use MetaProViz:
- To perform differential metabolite analysis (DMA) to generate Log2FC
and statistics and perform pathway analysis using Over Representation
Analysis (ORA) on the results.
- To do metabolite clustering analysis (MCA) to find clusters of
metabolites with similar behaviors based on patients demographics like
age, gender and tumour stage.
- Find the main metabolite drivers that separate patients based on
their demographics like age, gender and tumour stage.
First if you have not done yet, install the required dependencies and load the libraries:
# 1. Install Rtools if you haven’t done this yet, using the appropriate version (e.g.windows or macOS).
# 2. Install the latest development version from GitHub using devtools
#devtools::install_github("https://github.com/saezlab/MetaProViz")
library(MetaProViz)
#> Error in get(paste0(generic, ".", class), envir = get_method_env()) :
#> object 'type_sum.accel' not found
#dependencies that need to be loaded:
library(magrittr)
library(dplyr)
library(rlang)
library(tidyr)
library(tibble)
#Please install the Biocmanager Dependencies:
#BiocManager::install("clusterProfiler")
#BiocManager::install("EnhancedVolcano")1. Loading the example data
Here we choose an example datasets, which is publicly available in the
paper
“An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma”,
which includes metabolomic profiling on 138 matched clear cell renal
cell carcinoma (ccRCC)/normal tissue pairs (Hakimi et al. 2016). Metabolomics was done
using The company Metabolon, so this is untargeted metabolomics. Here we
use the median normalised data from the supplementary table 2 of the
paper. We have combined the metainformation about the patients with the
metabolite measurements and removed not identified metabolites. Lastly,
we have added a column “Stage” where Stage1 and Stage2 patients are
summarised to “EARLY-STAGE” and Stage3 and Stage4 patients to
“LATE-STAGE”. Moreover, we have added a column “Age”, where patients
with “AGE AT SURGERY” <42 are defined as “Young” and patients with
AGE AT SURGERY >58 as “Old” and the remaining patients as
“Middle”.
#As part of the
MetaProViz package you can load the example data into
your global environment using the function
toy_data():1. Tissue experiment (Intra)
We can load the ToyData, which includes columns with Sample
information and columns with the median normalised measured metabolite
integrated peaks.
# Load the example data:
Tissue_Norm <- MetaProViz::ToyData("Tissue_Norm")| TISSUE_TYPE | GENDER | AGE_AT_SURGERY | TYPE-STAGE | STAGE | AGE | 1,2-propanediol | 1,3-dihydroxyacetone | |
|---|---|---|---|---|---|---|---|---|
| DIAG-16076 | TUMOR | Male | 74.7778 | TUMOR-STAGE I | EARLY-STAGE | Old | 0.710920 | 0.809182 |
| DIAG-16077 | NORMAL | Male | 74.7778 | NORMAL-STAGE I | EARLY-STAGE | Old | 0.339390 | 0.718725 |
| DIAG-16078 | TUMOR | Male | 77.1778 | TUMOR-STAGE I | EARLY-STAGE | Old | 0.413386 | 0.276412 |
| DIAG-16079 | NORMAL | Male | 77.1778 | NORMAL-STAGE I | EARLY-STAGE | Old | 1.595697 | 4.332451 |
| DIAG-16080 | TUMOR | Female | 59.0889 | TUMOR-STAGE I | EARLY-STAGE | Old | 0.573787 | 0.646791 |
2. Additional information mapping the trivial metabolite
names to KEGG IDs, HMDB IDs, etc. and selected pathways
(MappingInfo)
Tissue_MetaData <- MetaProViz::ToyData("Tissue_MetaData")| SUPER_PATHWAY | SUB_PATHWAY | CAS | PUBCHEM | KEGG | Group_HMDB | |
|---|---|---|---|---|---|---|
| 1,2-propanediol | Lipid | Ketone bodies | 57-55-6; | NA | C00583 | HMDB01881 |
| 1,3-dihydroxyacetone | Carbohydrate | Glycolysis, gluconeogenesis, pyruvate metabolism | 62147-49-3; | 670 | C00184 | HMDB01882 |
| 1,5-anhydroglucitol (1,5-AG) | Carbohydrate | Glycolysis, gluconeogenesis, pyruvate metabolism | 154-58-5; | NA | C07326 | HMDB02712 |
| 10-heptadecenoate (17:1n7) | Lipid | Long chain fatty acid | 29743-97-3; | 5312435 | NA | NA |
| 10-nonadecenoate (19:1n9) | Lipid | Long chain fatty acid | 73033-09-7; | 5312513 | NA | NA |
2. Run MetaProViz Analysis
Pre-processing
This has been done by the authors of the paper and we will use the
median normalized data. If you want to know how you can use the
MetaProViz pre-processing module, please check out the
vignette:
- Standard
metabolomics data
- Consumption-Release
(CoRe) metabolomics data from cell culture media
Metadata analysis
We can use the patient’s metadata to find the main metabolite drivers
that separate patients based on their demographics like age, gender,
etc.
Here the metadata analysis is based on principal component analysis
(PCA), which is a dimensionality reduction method that reduces all the
measured features (=metabolites) of one sample into a few features in
the different principal components, whereby each principal component can
explain a certain percentage of the variance between the different
samples. Hence, this enables interpretation of sample clustering based
on the measured features (=metabolites).
The MetaProViz::MetaAnalysis() function will perform PCA to
extract the different PCs followed by annova to find the main metabolite
drivers that separate patients based on their demographics.
MetaRes <- MetaProViz::MetaAnalysis(InputData=Tissue_Norm[,-c(1:13)],
SettingsFile_Sample= Tissue_Norm[,c(2,4:5,12:13)],
Scaling = TRUE,
Percentage = 0.1,
StatCutoff= 0.05,
SaveAs_Table = "csv",
SaveAs_Plot = "svg",
PrintPlot= TRUE,
FolderPath = NULL)
#> The column names of the 'SettingsFile_Sample' contain special character that where removed.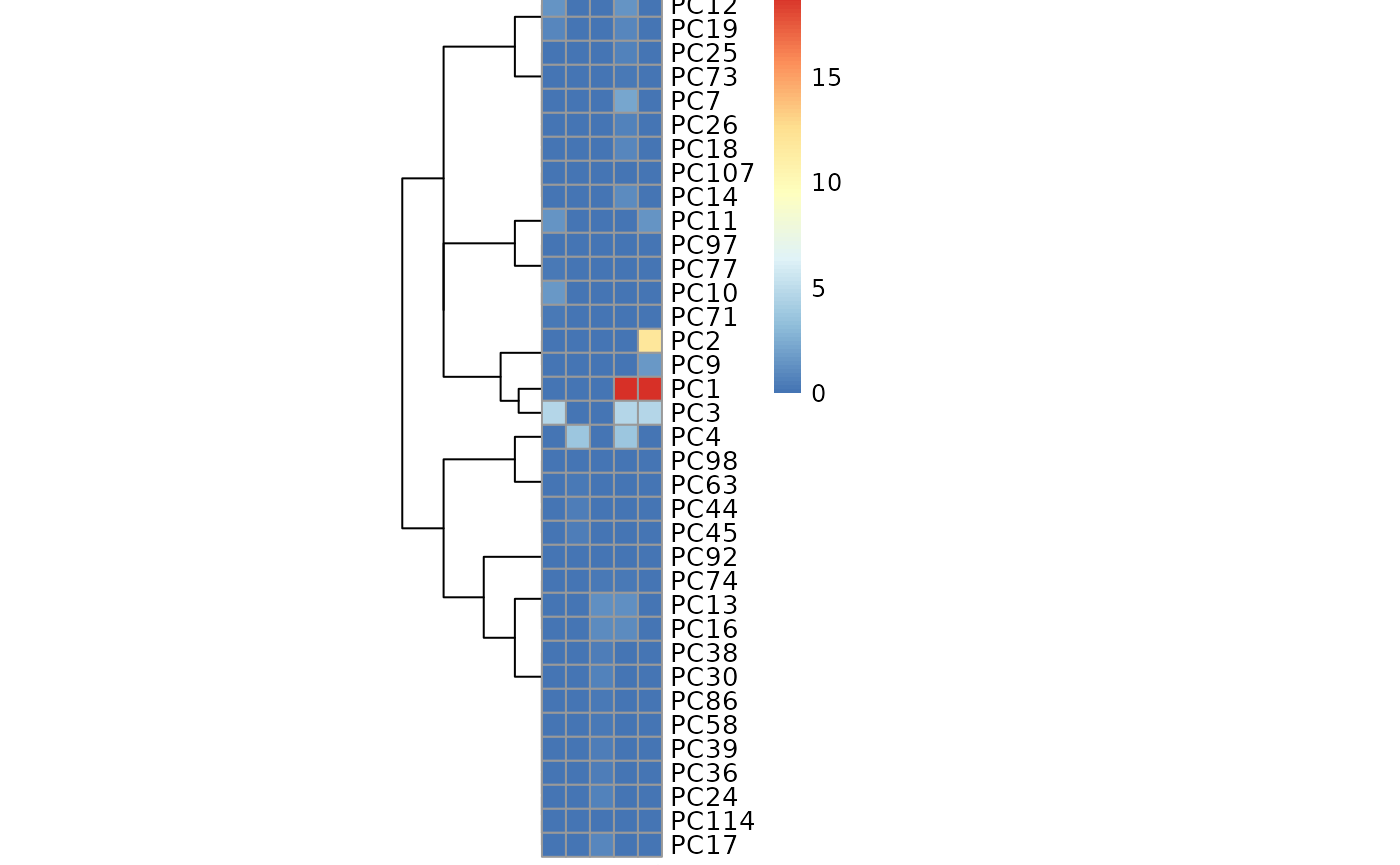
Ultimately, this is leading to clusters of metabolites that are driving the separation of the different demographics.
We generated the general anova output DF:
| PC | tukeyHSD_Contrast | term | anova_sumsq | anova_meansq | anova_statistic | anova_p.value | tukeyHSD_p.adjusted | Explained_Variance | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PC1 | TUMOR-NORMAL | TISSUE_TYPE | 7375.2212254 | 7375.2212254 | 90.1352800 | 0.0000000 | 0.0000000 | 19.0079573 |
| 2 | PC1 | Black-Asian | RACE | 156.7549451 | 52.2516484 | 0.4795311 | 0.6967838 | 0.9992824 | 19.0079573 |
| 1777 | PC232 | White-Asian | RACE | 0.2726952 | 0.0908984 | 0.9544580 | 0.4147472 | 0.5495634 | 0.0166997 |
| 1778 | PC232 | LATE-STAGE-EARLY-STAGE | STAGE | 0.0360319 | 0.0360319 | 0.3776760 | 0.5393596 | 0.5393596 | 0.0166997 |
| 3191 | PC9 | White-Other | RACE | 20.2626222 | 6.7542074 | 0.7090398 | 0.5473277 | 0.8897908 | 1.6658973 |
| 3192 | PC9 | Young-Middle | AGE | 49.2030973 | 24.6015486 | 2.6213834 | 0.0745317 | 0.9979475 | 1.6658973 |
We generated the summarised results output DF, where each feature (=metabolite) was assigned a main demographics parameter this feature is separating:
| FeatureID | term | Sum(Explained_Variance) | MainDriver | MainDriver_Term | MainDriver_Sum(VarianceExplained) |
|---|---|---|---|---|---|
| N2-methylguanosine | AGE, GENDER, RACE, STAGE, TISSUE_TYPE | 3.7598809871366, 2.75344828467363, 1.43932034747081, 25.280327361293, 33.9484968841434 | FALSE, FALSE, FALSE, FALSE, TRUE | TISSUE_TYPE | 33.9485 |
| 5-methyltetrahydrofolate (5MeTHF) | AGE, GENDER, RACE, STAGE, TISSUE_TYPE | 0.35114340819656, 0.29468807988021, 0.252515004077172, 19.4058160726611, 32.5442969233501 | FALSE, FALSE, FALSE, FALSE, TRUE | TISSUE_TYPE | 32.5443 |
| N-acetylalanine | AGE, GENDER, RACE, STAGE, TISSUE_TYPE | 0.381811888038144, 1.82507161869685, 2.97460134435356, 19.0079573465895, 32.5442969233501 | FALSE, FALSE, FALSE, FALSE, TRUE | TISSUE_TYPE | 32.5443 |
| N-acetyl-aspartyl-glutamate (NAAG) | AGE, GENDER, RACE, STAGE, TISSUE_TYPE | 0.212976478603115, 2.75344828467363, 0.235952044704099, 20.3572056704699, 32.2825995362096 | FALSE, FALSE, FALSE, FALSE, TRUE | TISSUE_TYPE | 32.2826 |
| 1-heptadecanoylglycerophosphoethanolamine* | AGE, GENDER, RACE, STAGE, TISSUE_TYPE | 4.33031140898824, 0.26785101869799, 0.437853800763463, 22.8424358182214, 30.8783995754163 | FALSE, FALSE, FALSE, FALSE, TRUE | TISSUE_TYPE | 30.8784 |
| 1-linoleoylglycerophosphoethanolamine* | AGE, STAGE, TISSUE_TYPE | 4.20115004304429, 22.7555733683955, 30.8783995754163 | FALSE, FALSE, TRUE | TISSUE_TYPE | 30.8784 |
##1. Tissue_Type
TissueTypeList <- MetaRes[["res_summary"]]%>%
filter(MainDriver_Term == "TISSUE_TYPE")%>%
filter(`MainDriver_Sum(VarianceExplained)`>30)%>%
select(FeatureID)%>%
pull()
#select columns Tissue_norm that are in TissueTypeList if they exist
Input_Heatmap <- Tissue_Norm[ , names(Tissue_Norm) %in% TissueTypeList]#c("N1-methylguanosine", "N-acetylalanine", "lysylmethionine")
#Heatmap: Metabolites that separate the demographics, like here TISSUE_TYPE
MetaProViz:::VizHeatmap(InputData = Input_Heatmap,
SettingsFile_Sample = Tissue_Norm[,c(1:13)],
SettingsInfo = c(color_Sample = list("TISSUE_TYPE")),
Scale ="column",
PlotName = "MainDrivers")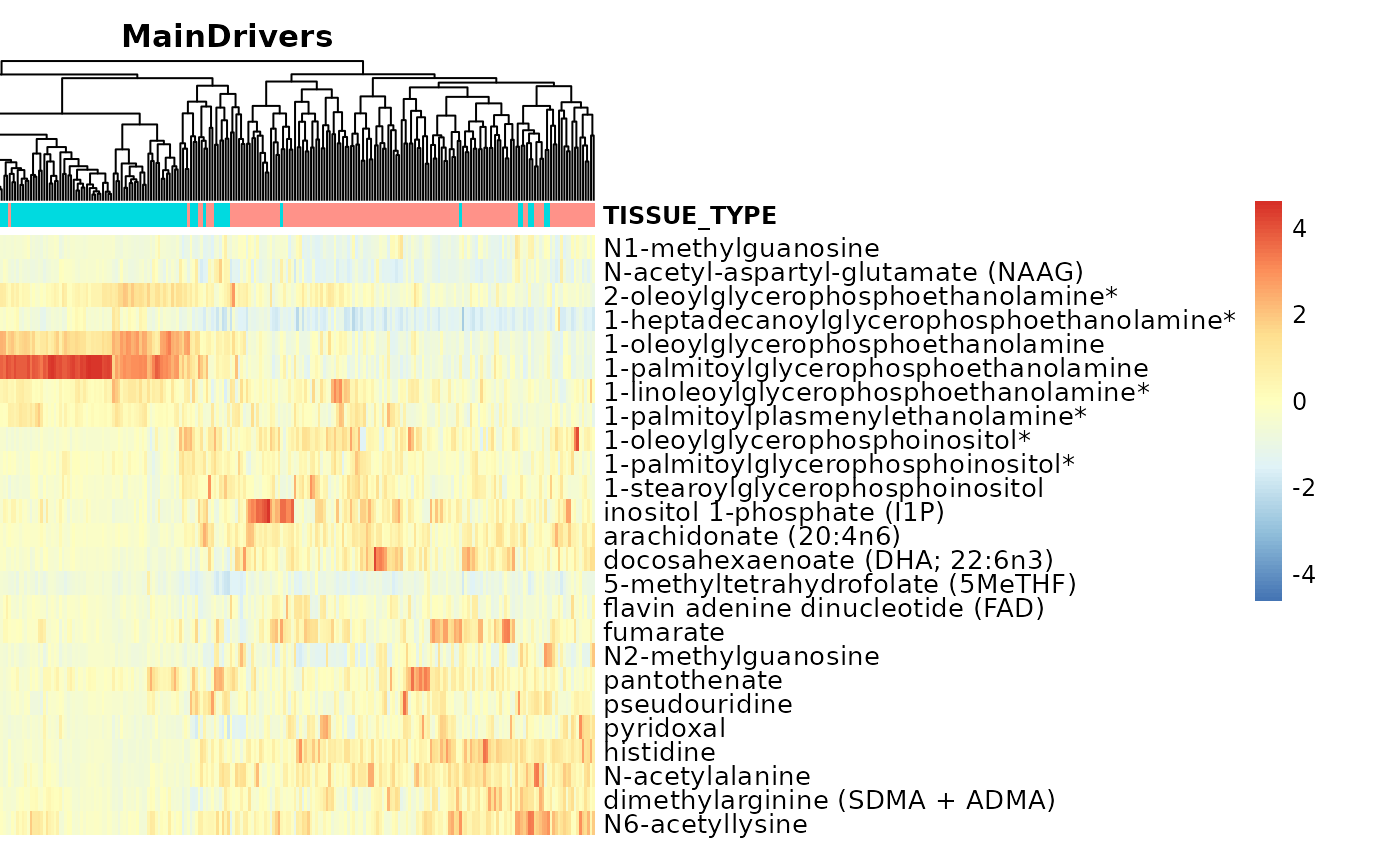
DMA
Here we use Differential Metabolite Analysis (DMA) to
compare two conditions (e.g. Tumour versus Healthy) by calculating the
Log2FC, p-value, adjusted p-value and t-value.
For more information please see the vignette:
- Standard
metabolomics data
- Consumption-Release
(CoRe) metabolomics data from cell culture media
We will perform multiple comparisons based on the different patient
demographics available: 1. Tumour versus Normal: All patients 2. Tumour
versus Normal: Subset of Early Stage patients 3. Tumour
versus Normal: Subset of Late Stage patients 4. Tumour
versus Normal: Subset of Young patients 5. Tumour versus
Normal: Subset of Old patients
#Prepare the different selections
EarlyStage <- Tissue_Norm%>%
filter(STAGE== "EARLY-STAGE")
LateStage <- Tissue_Norm%>%
filter(STAGE=="LATE-STAGE")
Old <- Tissue_Norm%>%
filter(AGE=="Old")
Young <- Tissue_Norm%>%
filter(AGE=="Young")
DFs <- list("TissueType"= Tissue_Norm,"EarlyStage"= EarlyStage, "LateStage"= LateStage, "Old"= Old, "Young"=Young)
#Run DMA
ResList <- list()
for(item in names(DFs)){
#Get the right DF:
InputData <- DFs[[item]]
#Perform DMA
message(paste("Running DMA for", item))
TvN <- MetaProViz::DMA(InputData = InputData[,-c(1:13)],
SettingsFile_Sample = InputData[,c(1:13)],
SettingsInfo = c(Conditions="TISSUE_TYPE", Numerator="TUMOR" , Denominator = "NORMAL"),
PerformShapiro=FALSE) #The data have been normalized by the company that provided the results and include metabolites with zero variance as they were all imputed with the same missing value.
#Add Results to list
ResList[[item]] <- TvN
}#> Running DMA for TissueType
#> There are no NA/0 values
#> Running DMA for EarlyStage
#> There are no NA/0 values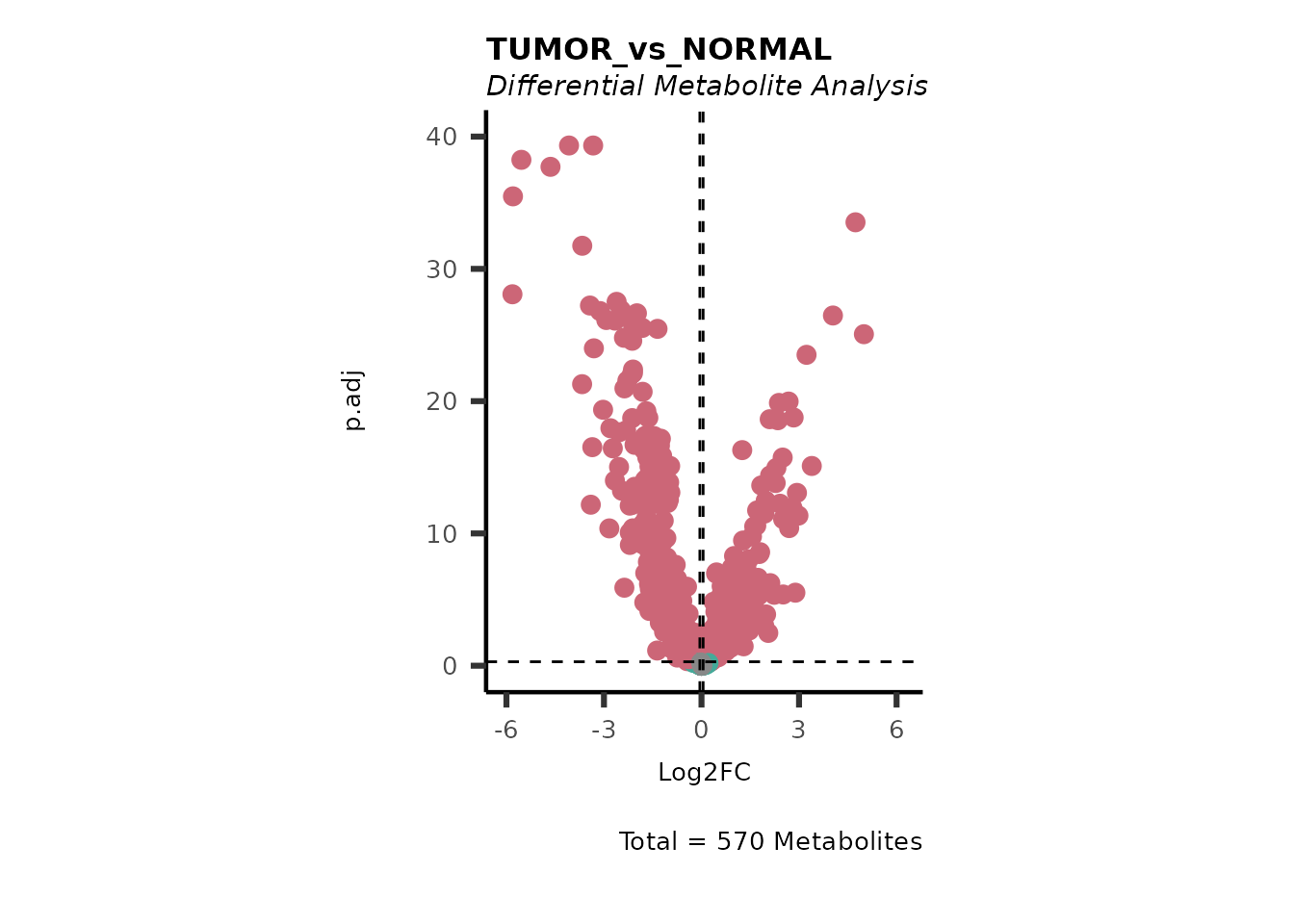
#> Running DMA for LateStage
#> There are no NA/0 values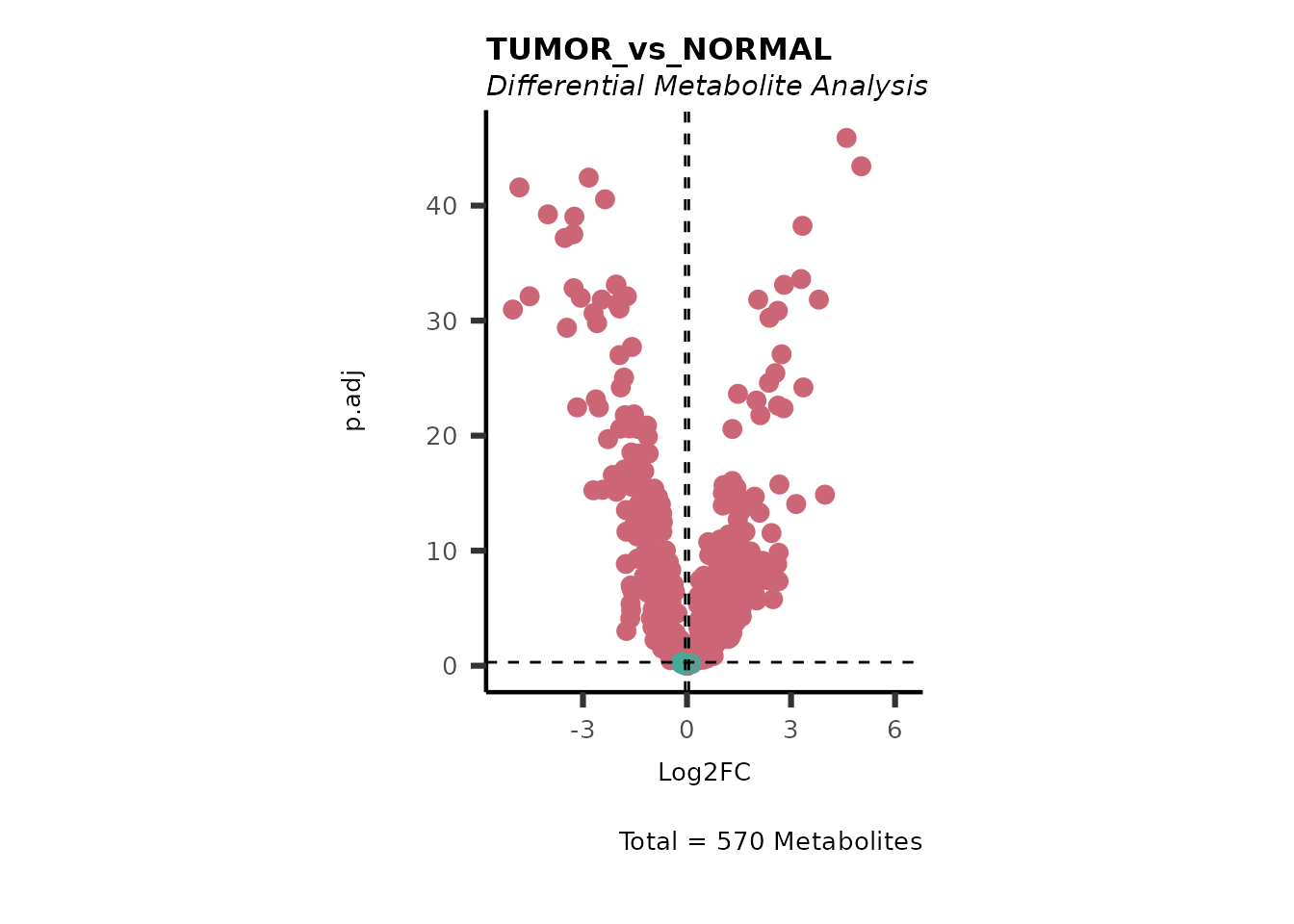
#> Running DMA for Old
#> There are no NA/0 values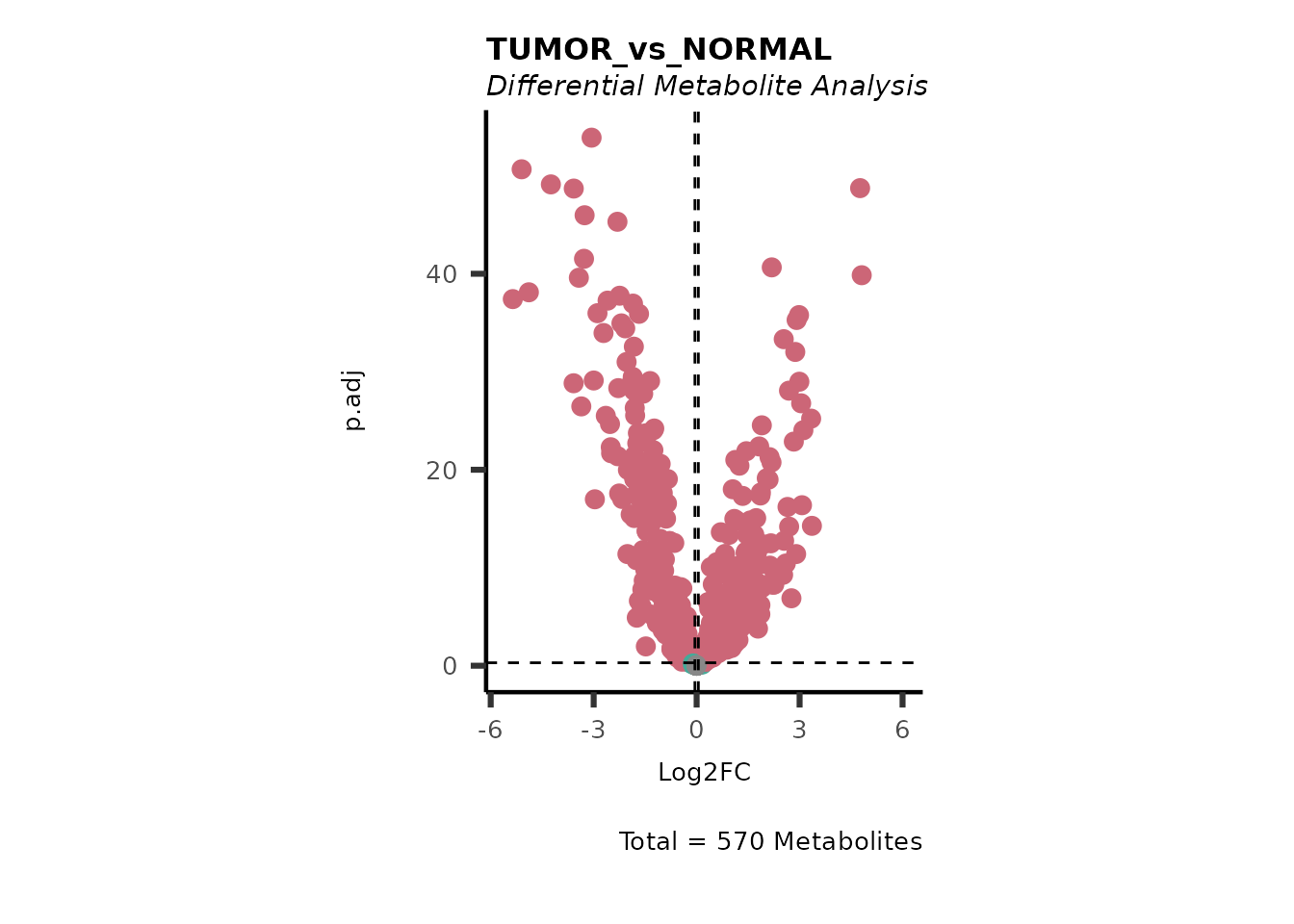
#> Running DMA for Young
#> There are no NA/0 values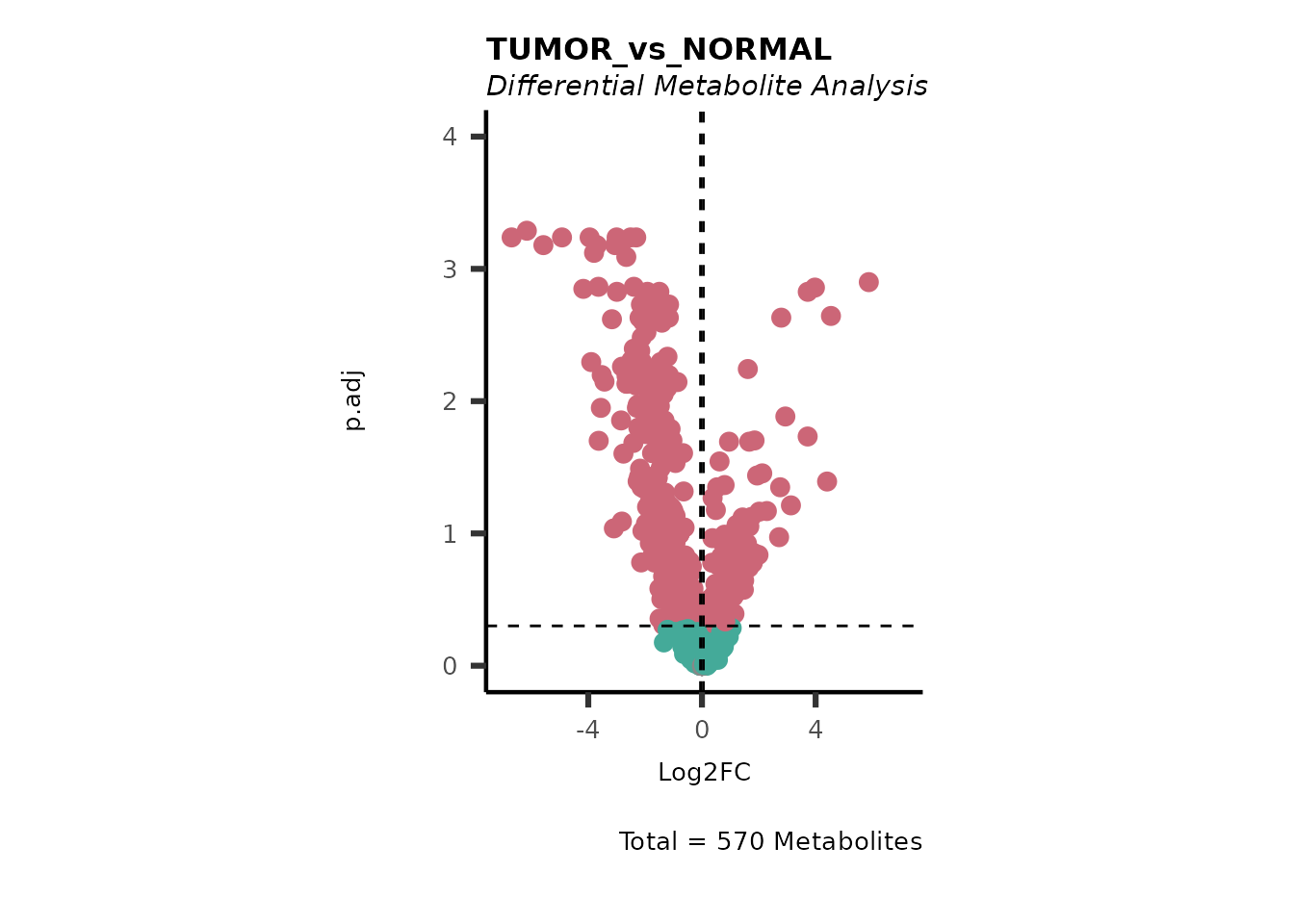
We can see from the different Volcano plots have smaller p.adjusted
values and differences in Log2FC range.
Here we can also use the MetaproViz::VizVolcano() function
to plot comparisons together on the same plot, such as Tumour versus
Normal of young and old patients:
#Early versus Late Stage
MetaProViz::VizVolcano(PlotSettings="Compare",
InputData=ResList[["EarlyStage"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
InputData2= ResList[["LateStage"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
ComparisonName= c(InputData="EarlyStage", InputData2= "LateStage"),
PlotName= "EarlyStage-TUMOR_vs_NORMAL compared to LateStage-TUMOR_vs_NORMAL",
Subtitle= "Results of DMA" )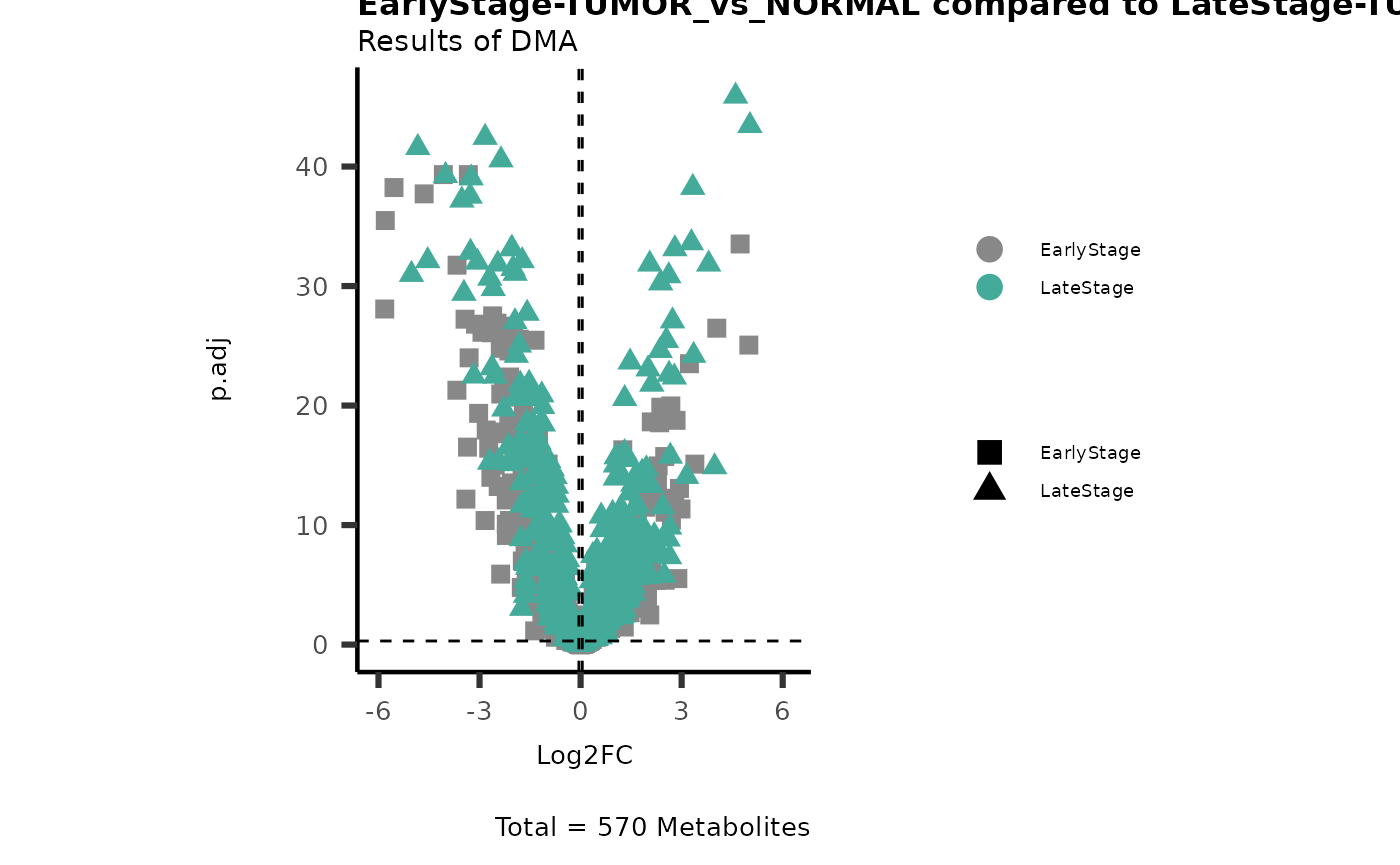
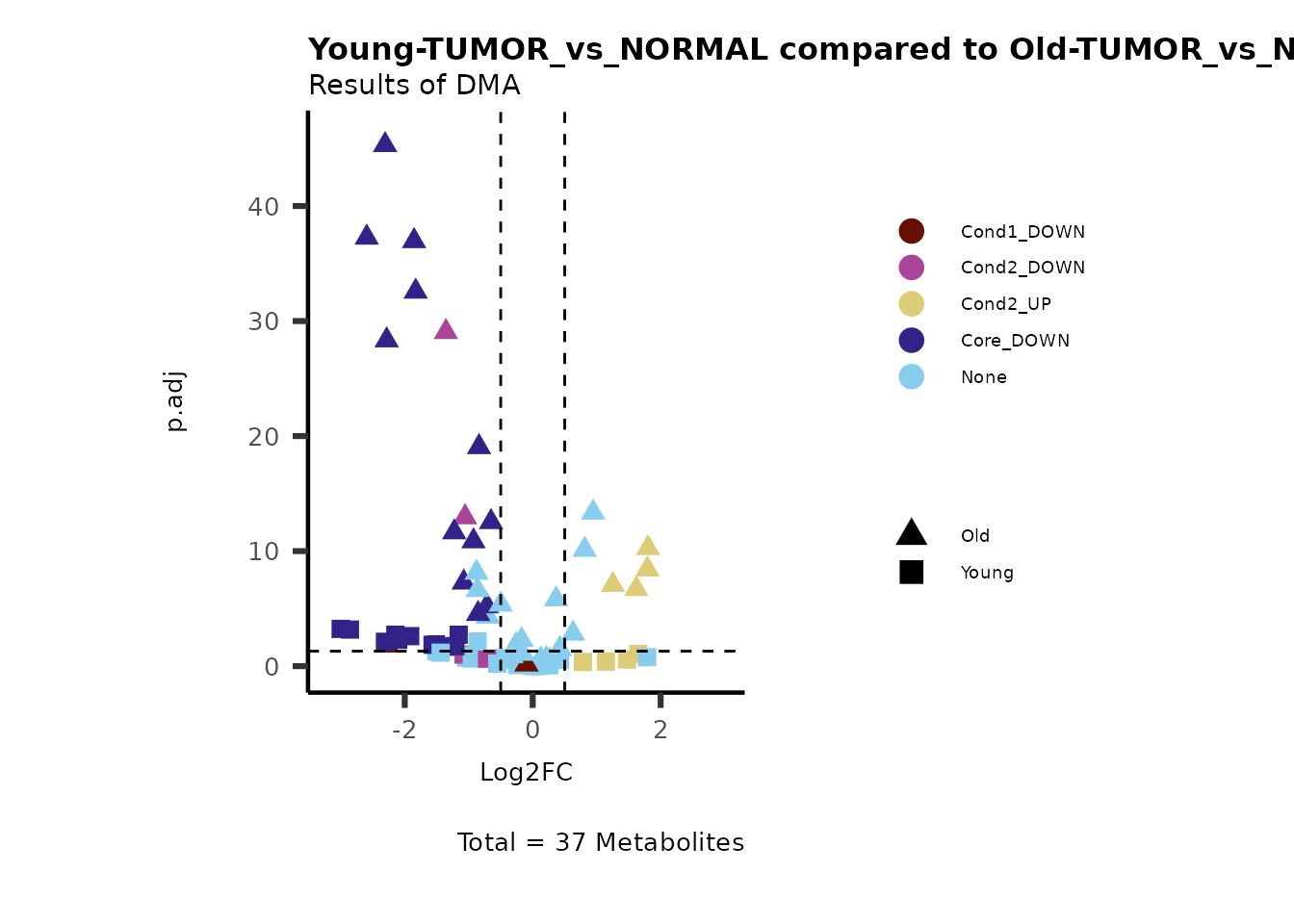
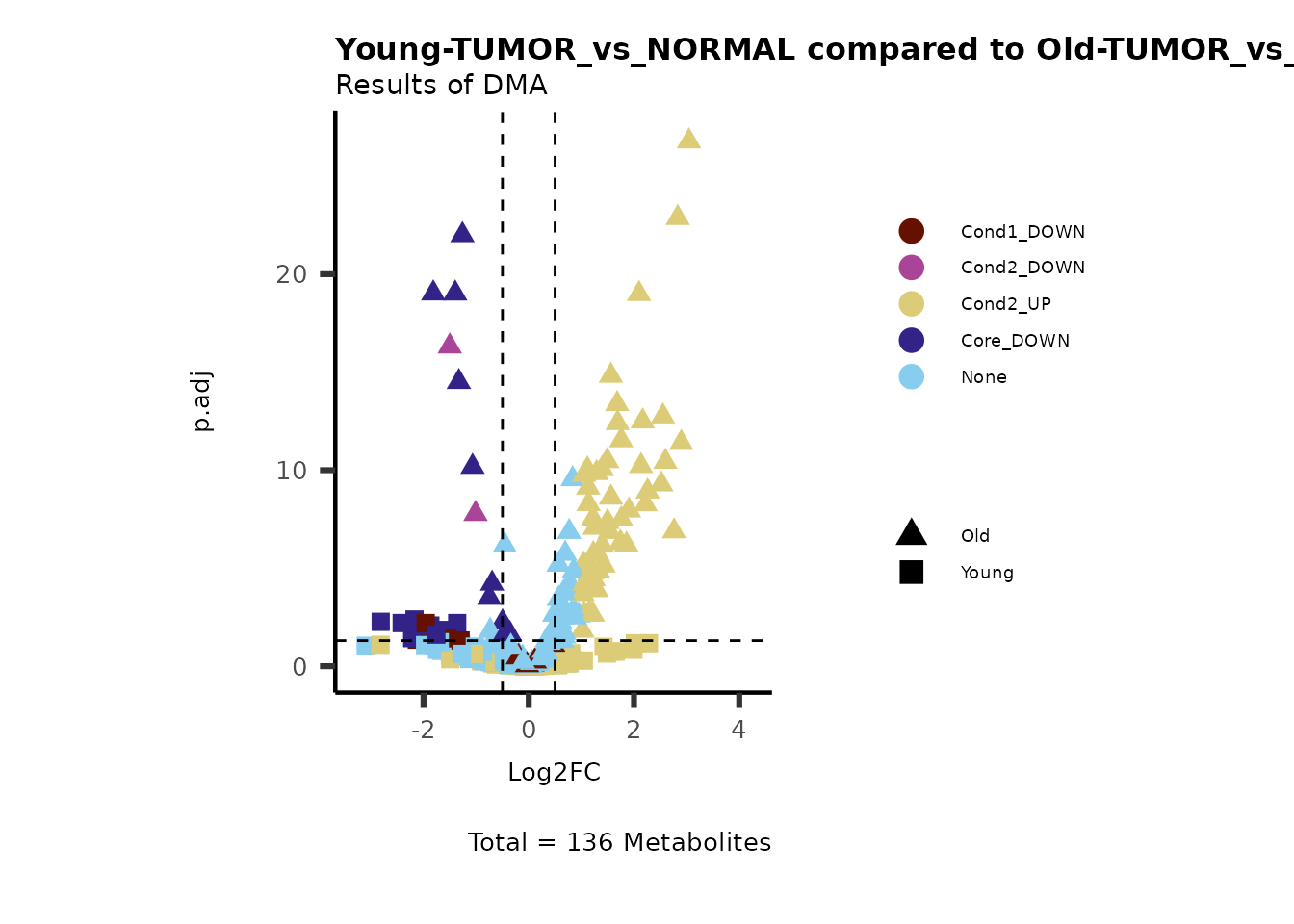
# Young versus Old
MetaProViz::VizVolcano(PlotSettings="Compare",
InputData=ResList[["Young"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
InputData2= ResList[["Old"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
ComparisonName= c(InputData="Young", InputData2= "Old"),
PlotName= "Young-TUMOR_vs_NORMAL compared to Old-TUMOR_vs_NORMAL",
Subtitle= "Results of DMA" )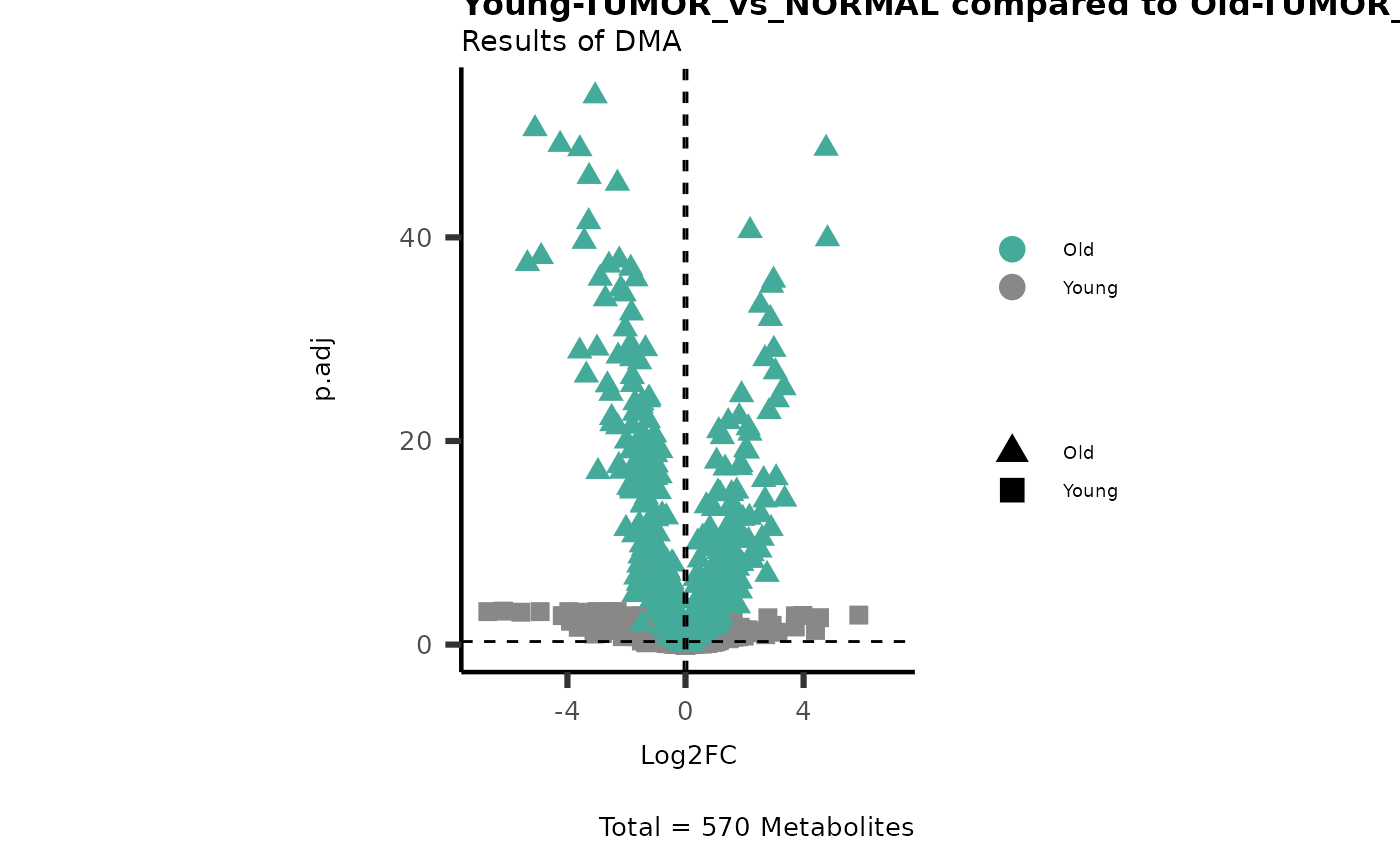
Here we can observe that Tumour versus Normal has lower significance
values for the Young patients compared to the Old patients. This can be
due to higher variance in the metabolite measurements from Young
patients compared to the Old patients.
Lastly, we can also check if the top changed metabolites comparing
Tumour versus Normal correlate with the main metabolite drivers that
separate patients based on their TISSUE_TYPE, which are
Tumour or Normal.
#Get the top changed metabolites
top_entries <- ResList[["TissueType"]][["DMA"]][["TUMOR_vs_NORMAL"]] %>%
arrange(desc(t.val)) %>%
slice(1:25)%>%
select(Metabolite)%>%
pull()
bottom_entries <- ResList[["TissueType"]][["DMA"]][["TUMOR_vs_NORMAL"]] %>%
arrange(desc(t.val)) %>%
slice((n()-24):n())%>%
select(Metabolite) %>%
pull()
#Check if those overlap with the top demographics drivers
ggVennDiagram::ggVennDiagram(list(Top = top_entries,
Bottom = bottom_entries,
TissueTypeList = TissueTypeList))+
scale_fill_gradient(low = "blue", high = "red")
MetaData_Metab <- merge(x=Tissue_MetaData,
y= MetaRes[["res_summary"]][, c(1,5:6) ]%>%tibble::column_to_rownames("FeatureID"),
by=0,
all.y=TRUE)%>%
column_to_rownames("Row.names")
#Make a Volcano plot:
MetaProViz::VizVolcano(PlotSettings="Standard",
InputData=ResList[["TissueType"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
SettingsFile_Metab = MetaData_Metab,
SettingsInfo = c(color = "MainDriver_Term"),
PlotName= "TISSUE_TYPE-TUMOR_vs_NORMAL",
Subtitle= "Results of DMA" )
Biological regulated clustering
To understand which metabolites are changing independent of the
patients age, hence only due to tumour versus normal, and which
metabolites change independent of tumour versus normal, hence due to the
different age, we can use the MetaProViz::MCA_2Cond()
function.
Metabolite Clustering Analysis (MCA) enables clustering of
metabolites into groups based on logical regulatory rules. Here we set
two different thresholds, one for the differential metabolite abundance
(Log2FC) and one for the significance (e.g. p.adj). This will define if
a feature (= metabolite) is assigned into:
1. “UP”, which means a metabolite is significantly up-regulated in the
underlying comparison.
2. “DOWN”, which means a metabolite is significantly down-regulated in
the underlying comparison.
3. “No Change”, which means a metabolite does not change significantly
in the underlying comparison and/or is not defined as
up-regulated/down-regulated based on the Log2FC threshold chosen.
Thereby “No Change” is further subdivided into four states:
1. “Not Detected”, which means a metabolite is not detected in the
underlying comparison.
2. “Not Significant”, which means a metabolite is not significant in the
underlying comparison.
3. “Significant positive”, which means a metabolite is significant in
the underlying comparison and the differential metabolite abundance is
positive, yet does not meet the threshold set for “UP” (e.g. Log2FC
>1 = “UP” and we have a significant Log2FC=0.8).
4. “Significant negative”, which means a metabolite is significant in
the underlying comparison and the differential metabolite abundance is
negative, yet does not meet the threshold set for “DOWN”.
For more information you can also check out the other vignettes.
MCAres <- MetaProViz::MCA_2Cond(InputData_C1=ResList[["Young"]][["DMA"]][["TUMOR_vs_NORMAL"]],
InputData_C2=ResList[["Old"]][["DMA"]][["TUMOR_vs_NORMAL"]],
SettingsInfo_C1=c(ValueCol="Log2FC",StatCol="p.adj", StatCutoff= 0.05, ValueCutoff=1),
SettingsInfo_C2=c(ValueCol="Log2FC",StatCol="p.adj", StatCutoff= 0.05, ValueCutoff=1),
FeatureID = "Metabolite",
SaveAs_Table = "csv",
BackgroundMethod="C1&C2"#Most stringend background setting, only includes metabolites detected in both comparisons
)
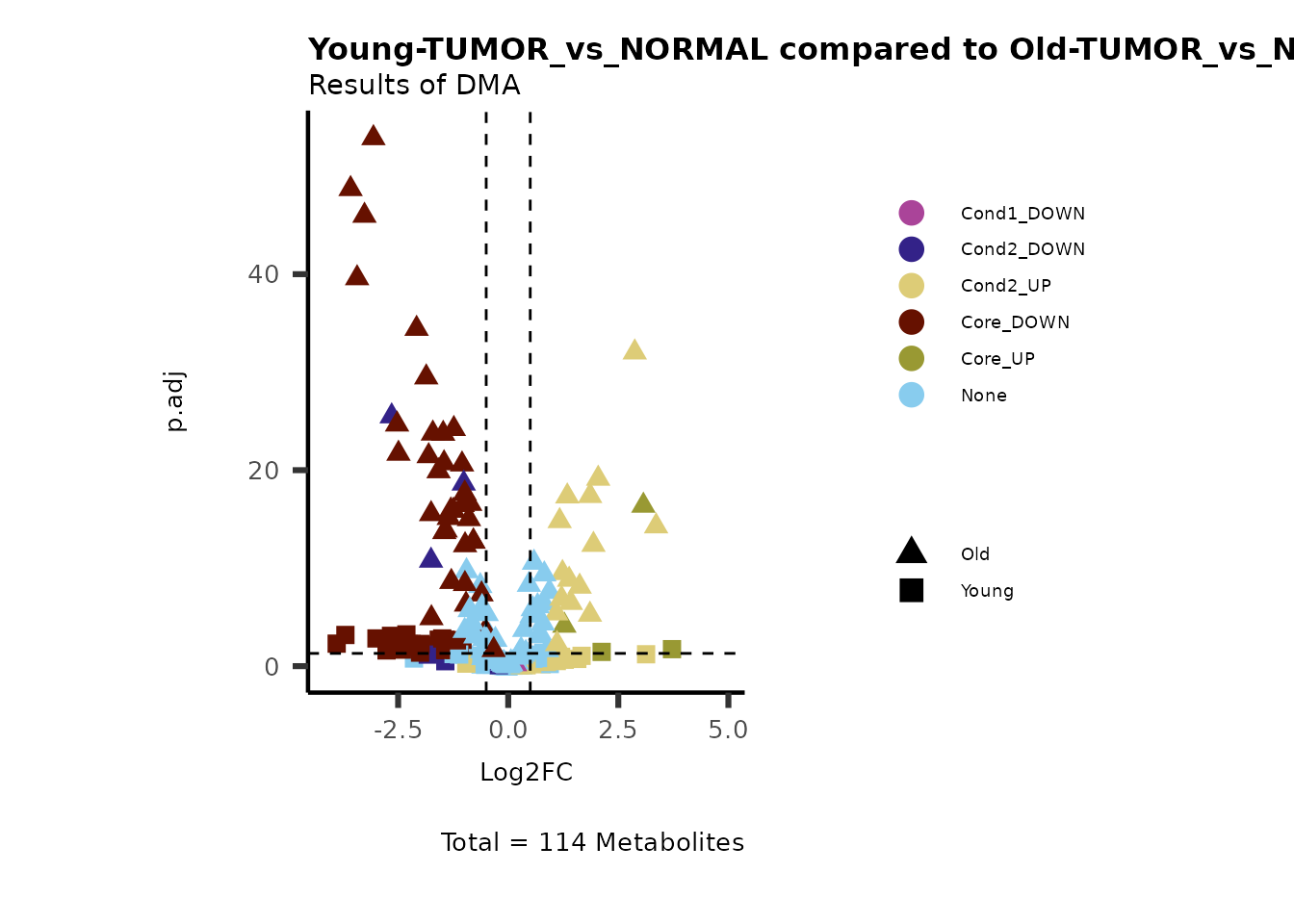
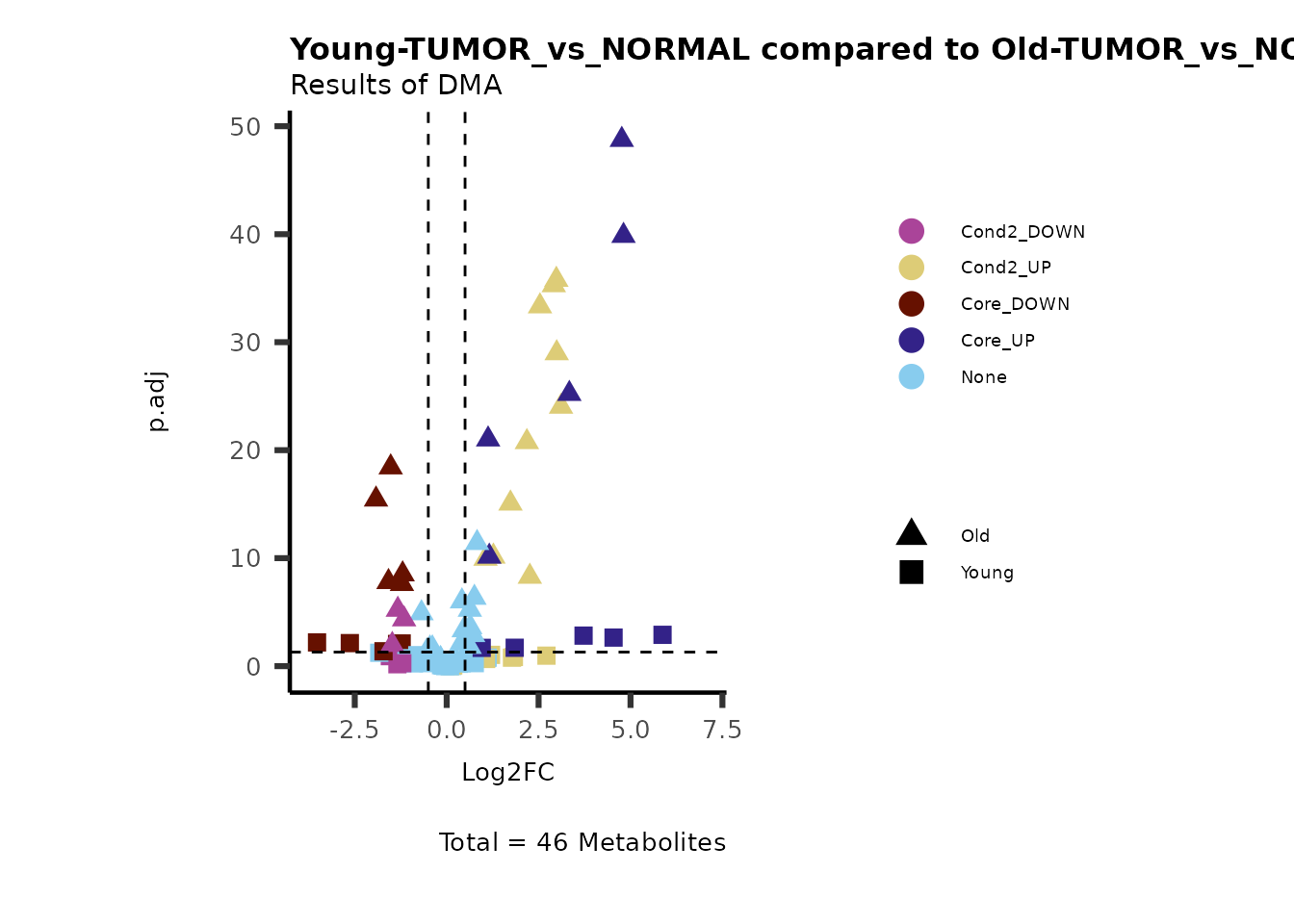
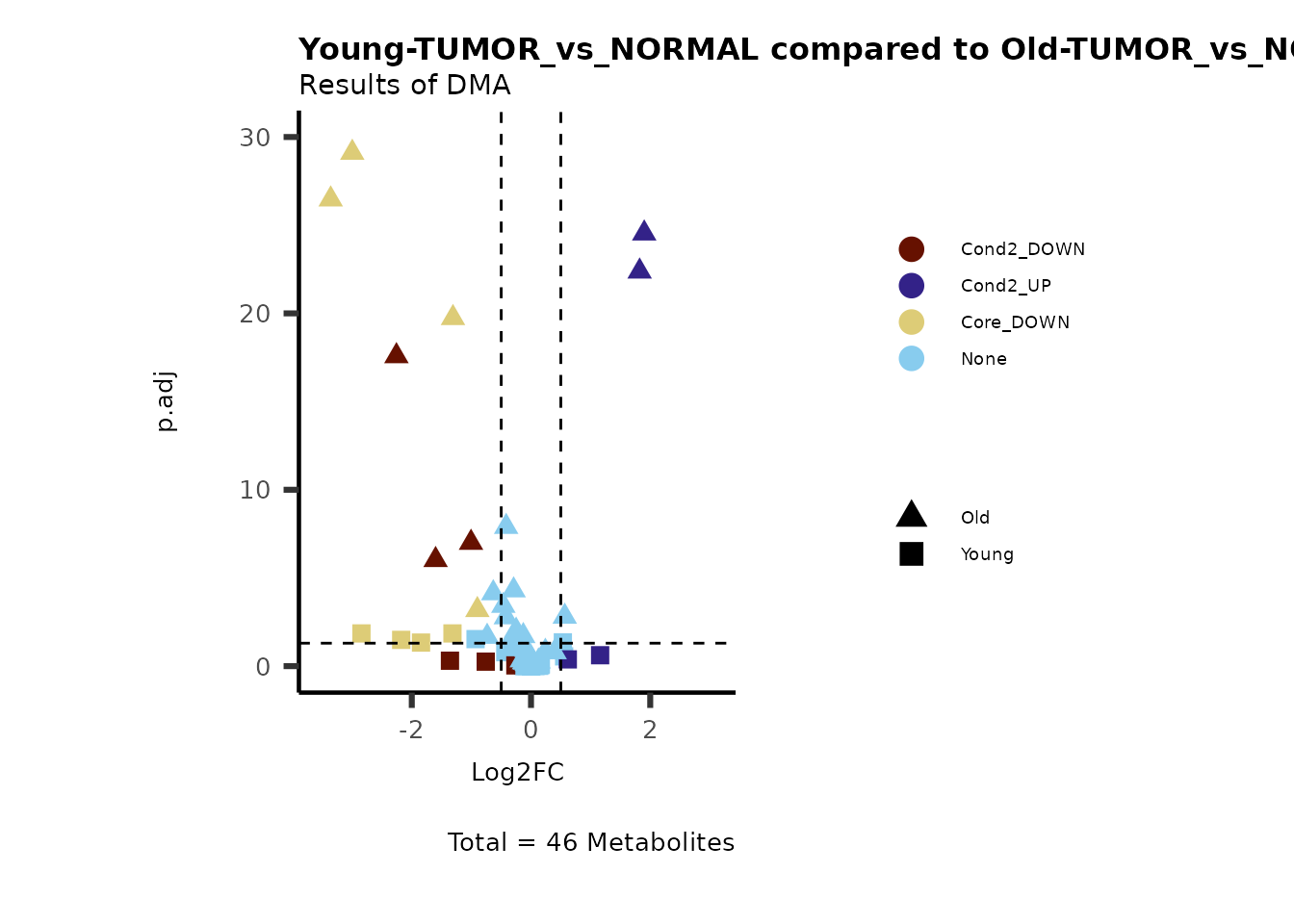
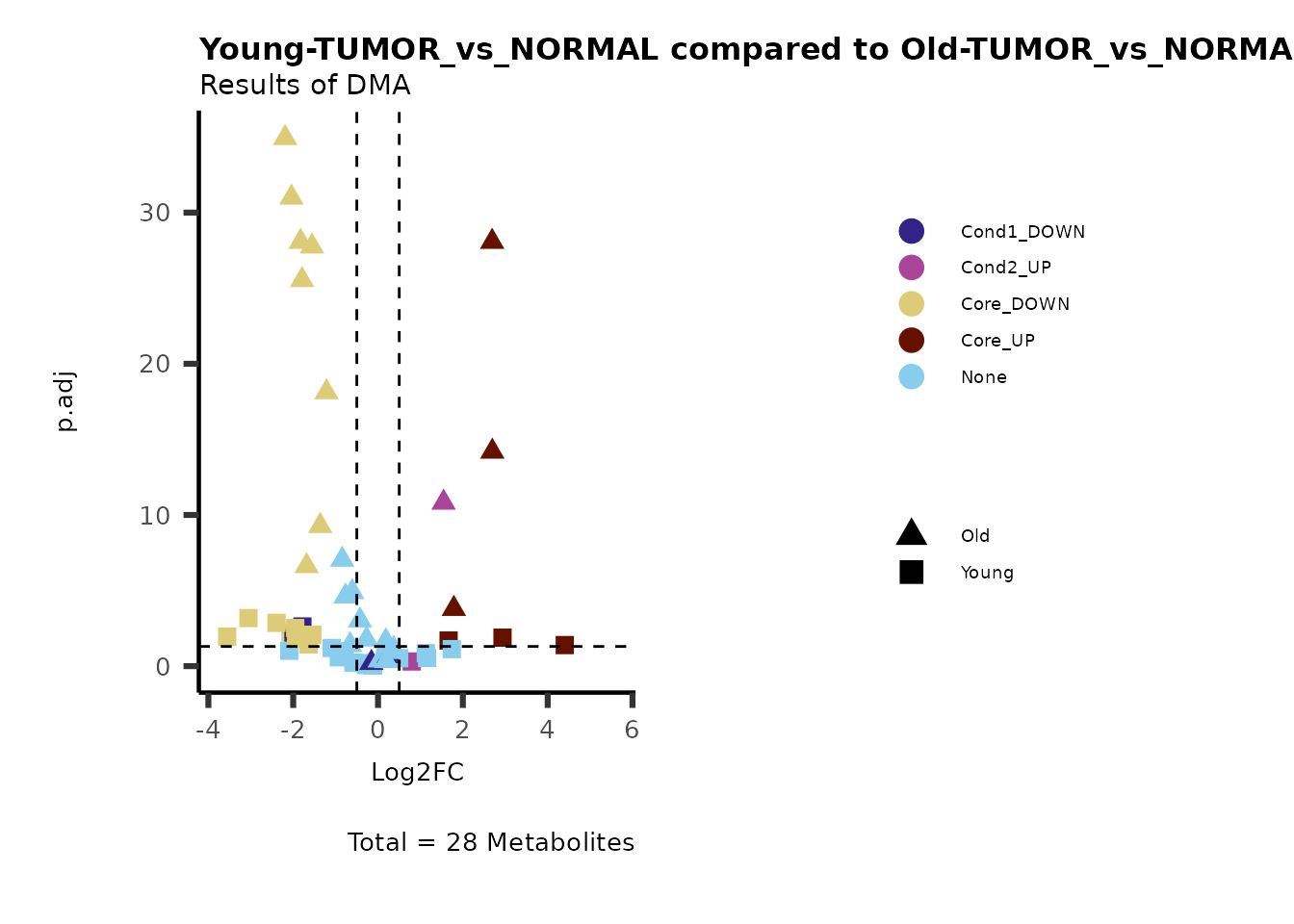
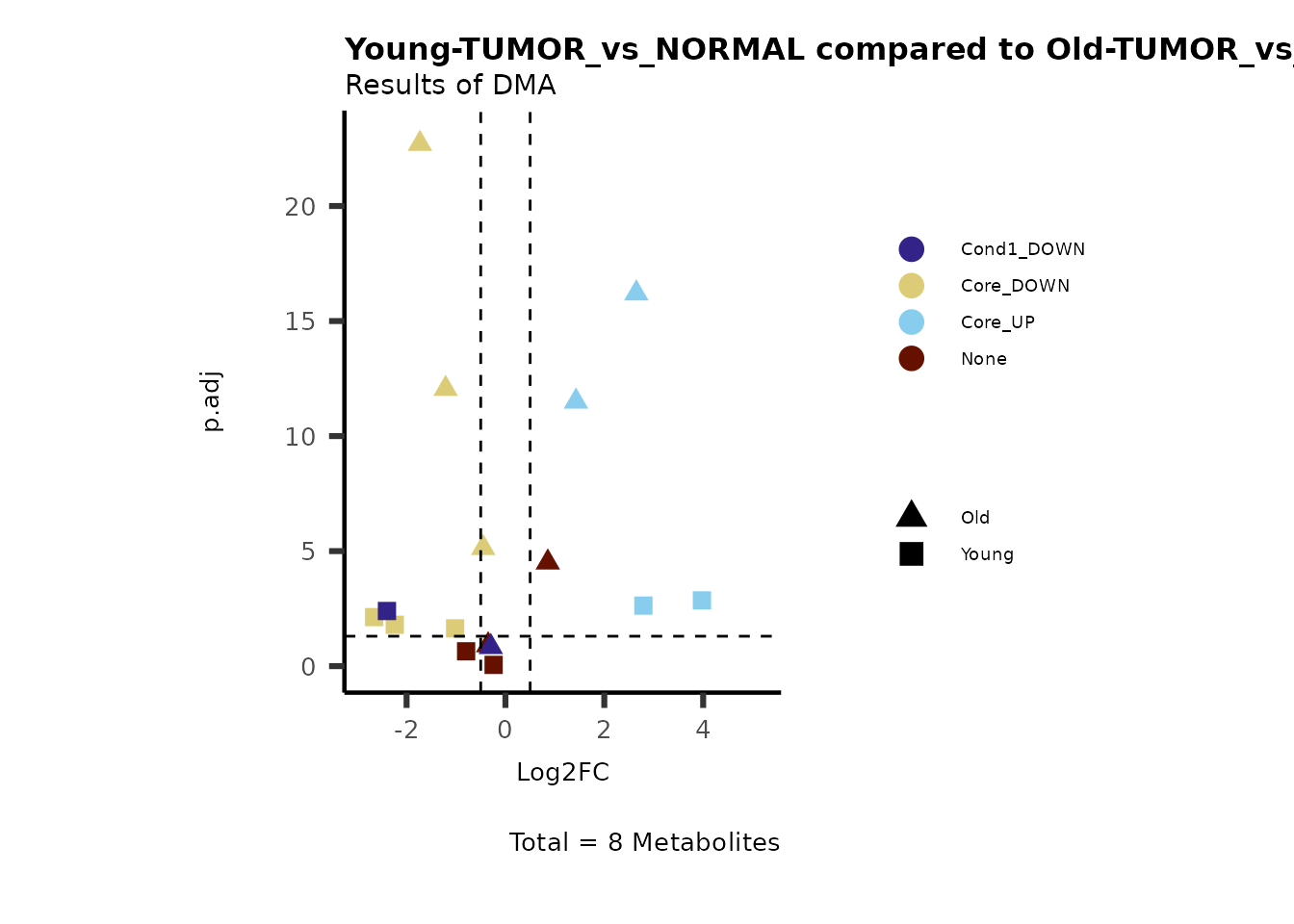
Now we can use this information to colour code our volcano plot:
#Add metabolite information such as KEGG ID or pathway to results
MetaData_Metab <- merge(x=Tissue_MetaData,
y= MCAres[["MCA_2Cond_Results"]][, c(1, 14:15)]%>%tibble::column_to_rownames("Metabolite"),
by=0,
all.y=TRUE)%>%
tibble::column_to_rownames("Row.names")
MetaProViz::VizVolcano(PlotSettings="Compare",
InputData=ResList[["Young"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
InputData2= ResList[["Old"]][["DMA"]][["TUMOR_vs_NORMAL"]]%>%tibble::column_to_rownames("Metabolite"),
ComparisonName= c(InputData="Young", InputData2= "Old"),
SettingsFile_Metab = MetaData_Metab,
PlotName= "Young-TUMOR_vs_NORMAL compared to Old-TUMOR_vs_NORMAL",
Subtitle= "Results of DMA",
SettingsInfo = c(individual = "SUPER_PATHWAY",
color = "RG2_Significant"))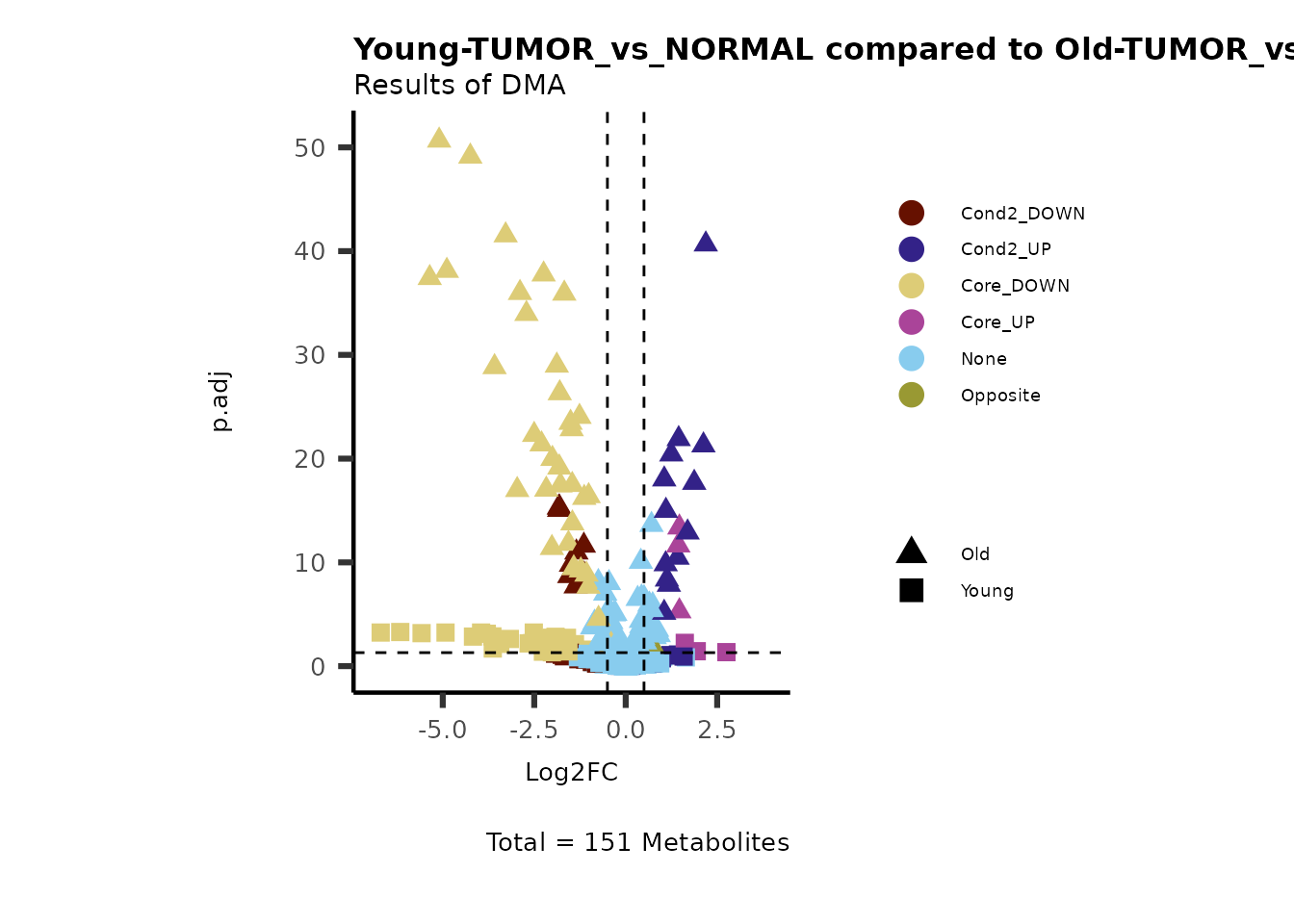
Session information
#> R version 4.4.2 (2024-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.1 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8
#> [6] LC_MESSAGES=C.UTF-8 LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] tibble_3.2.1 tidyr_1.3.1 rlang_1.1.4 dplyr_1.1.4 magrittr_2.0.3 MetaProViz_2.1.3
#> [7] ggplot2_3.5.1
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.2.3 gridExtra_2.3 logger_0.4.0 readxl_1.4.3 compiler_4.4.2
#> [6] RSQLite_2.3.9 systemfonts_1.1.0 vctrs_0.6.5 reshape2_1.4.4 rvest_1.0.4
#> [11] stringr_1.5.1 pkgconfig_2.0.3 crayon_1.5.3 fastmap_1.2.0 backports_1.5.0
#> [16] labeling_0.4.3 rmarkdown_2.29 tzdb_0.4.0 ggbeeswarm_0.7.2 ragg_1.3.3
#> [21] purrr_1.0.2 bit_4.5.0.1 xfun_0.49 cachem_1.1.0 jsonlite_1.8.9
#> [26] progress_1.2.3 blob_1.2.4 later_1.4.1 broom_1.0.7 parallel_4.4.2
#> [31] prettyunits_1.2.0 R6_2.5.1 RColorBrewer_1.1-3 bslib_0.8.0 stringi_1.8.4
#> [36] limma_3.58.1 car_3.1-3 lubridate_1.9.4 jquerylib_0.1.4 cellranger_1.1.0
#> [41] Rcpp_1.0.13-1 knitr_1.49 R.utils_2.12.3 readr_2.1.5 igraph_2.1.2
#> [46] timechange_0.3.0 tidyselect_1.2.1 rstudioapi_0.17.1 abind_1.4-8 yaml_2.3.10
#> [51] ggVennDiagram_1.5.2 curl_6.0.1 plyr_1.8.9 withr_3.0.2 inflection_1.3.6
#> [56] evaluate_1.0.1 desc_1.4.3 zip_2.3.1 xml2_1.3.6 pillar_1.10.0
#> [61] ggpubr_0.6.0 carData_3.0-5 checkmate_2.3.2 generics_0.1.3 vroom_1.6.5
#> [66] hms_1.1.3 munsell_0.5.1 scales_1.3.0 gtools_3.9.5 OmnipathR_3.15.2
#> [71] glue_1.8.0 pheatmap_1.0.12 tools_4.4.2 ggsignif_0.6.4 fs_1.6.5
#> [76] XML_3.99-0.17 grid_4.4.2 qcc_2.7 colorspace_2.1-1 patchwork_1.3.0
#> [81] beeswarm_0.4.0 vipor_0.4.7 Formula_1.2-5 cli_3.6.3 rappdirs_0.3.3
#> [86] kableExtra_1.4.0 textshaping_0.4.1 viridisLite_0.4.2 svglite_2.1.3 gtable_0.3.6
#> [91] R.methodsS3_1.8.2 rstatix_0.7.2 hash_2.2.6.3 EnhancedVolcano_1.20.0 sass_0.4.9
#> [96] digest_0.6.37 ggrepel_0.9.6 htmlwidgets_1.6.4 farver_2.1.2 memoise_2.0.1
#> [101] htmltools_0.5.8.1 pkgdown_2.1.1 R.oo_1.27.0 factoextra_1.0.7 lifecycle_1.0.4
#> [106] httr_1.4.7 statmod_1.5.0 bit64_4.5.2 MASS_7.3-61