CoRe Metabolomics
Christina Schmidt
Heidelberg UniversityDimitrios Prymidis
Cologne UniversitySource:
vignettes/CoRe Metabolomics.Rmd
CoRe Metabolomics.Rmd
A Consumption-Release (CoRe) metabolomics experiment usually refers to a
cell culture experiment where metabolomics is performed on the cell
culture media.
In this tutorial we showcase how
to use MetaProViz:
- To process raw peak data and identify outliers.
- To perform differential metabolite analysis (DMA) to generate
Log2Distance and statistics and perform pathway analysis using Over
Representation Analysis (ORA) on the results.
- To do metabolite clustering analysis (MCA) to find clusters of
metabolites with similar behaviors and perform pathway analysis using
ORA on each cluster.
- To use specific visualizations to aid biological interpretation of
the results.
First if you have not done yet, install the required dependencies and load the libraries:
# 1. Install Rtools if you haven’t done this yet, using the appropriate version (e.g.windows or macOS).
# 2. Install the latest development version from GitHub using devtools
#devtools::install_github("https://github.com/saezlab/MetaProViz")
library(MetaProViz)
#> Error in get(paste0(generic, ".", class), envir = get_method_env()) :
#> object 'type_sum.accel' not found
#dependencies that need to be loaded:
library(magrittr)
library(dplyr)
library(tibble)
library(rlang)
library(ggfortify)
#Please install the Biocmanager Dependencies:
#BiocManager::install("clusterProfiler")
#BiocManager::install("EnhancedVolcano")1. Loading the example data
Here we choose an example datasets, which is publicly available on metabolomics
workbench project PR001418 including metabolic profiles of human
renal epithelial cells HK2 and cell renal cell carcinoma (ccRCC) cell
lines cultured in Plasmax cell culture media. Here we use the integrated
raw peak data as example data using the trivial metabolite name in
combination with the KEGG ID as the metabolite identifiers.
As part of the
MetaProViz package you can load the example data into
your global environment using the function
toy_data():1. CoRe experiment (CoRe)
The raw data are available via metabolomics
workbench study ST002226 were exometabolomics of HK2 and ccRCC cell
lines 786-O, 786-M1A, 786-M2A, OS-RC-2, OS-LM1 and RFX-631 were
performed.
Media <- MetaProViz::ToyData(Data="CultureMedia_Raw")| Conditions | Biological_Replicates | GrowthFactor | valine-d8 | hipppuric acid-d5 | 2-hydroxyglutarate | 2-ketoglutarate | 3-Dehydro-L-threonate | |
|---|---|---|---|---|---|---|---|---|
| MS51-06 | HK2 | 1 | 249.2817 | 780552871 | 3127630257 | 84950547 | 169244158 | 1807489245 |
| MS51-07 | HK2 | 2 | 249.2817 | 802602348 | 3256031922 | 60753859 | 151064767 | 695228424 |
| MS51-08 | HK2 | 3 | 249.2817 | 831984796 | 3308009345 | 73718363 | 171281531 | 791442407 |
| MS51-09 | HK2 | 4 | 249.2817 | 822744518 | 3209731571 | 65933166 | 112033043 | 315209589 |
| MS51-10 | HK2 | 5 | 249.2817 | 805565867 | 3297793480 | 68183576 | 170902744 | 615035216 |
| MS51-11 | 786-O | 1 | 297.3423 | 841873509 | 3418515398 | 75941661 | 215553005 | 501977089 |
| MS51-12 | 786-O | 2 | 297.3423 | 825462964 | 3218049751 | 62100210 | 195308040 | 484681099 |
2. Additional information mapping the trivial metabolite
names to KEGG IDs and selected pathways
(MappingInfo)
MappingInfo <- MetaProViz::ToyData(Data="Cells_MetaData")| HMDB | KEGG.ID | KEGGCompound | Pathway | |
|---|---|---|---|---|
| N-acetylaspartate | HMDB0000812 | C01042 | N-Acetyl-L-aspartate | Alanine, aspartate and glutamate metabolism |
| argininosuccinate | HMDB0000052 | C03406 | N-(L-Arginino)succinate | Alanine, aspartate and glutamate metabolism |
| N-acetylaspartylglutamate | HMDB0001067 | C12270 | N-Acetylaspartylglutamate | Alanine, aspartate and glutamate metabolism |
| tyrosine | HMDB0000158 | C00082 | L-Tyrosine | Amino acid metabolism |
| asparagine | HMDB0000168 | C00152 | L-Asparagine | Amino acid metabolism |
3. KEGG pathways that are loaded via KEGG API using the
package KEGGREST and can be used to perform pathway
analysis. (KEGG_Pathways)
#This will use KEGGREST to query the KEGG API to load the pathways:
MetaProViz::LoadKEGG()
#> Cached file loaded from: ~/.cache/KEGG_Metabolite.rds| term | Metabolite | MetaboliteID | Description |
|---|---|---|---|
| Glycolysis / Gluconeogenesis - Homo sapiens (human) | Pyruvate | C00022 | Glycolysis / Gluconeogenesis - Homo sapiens (human) |
| Glycolysis / Gluconeogenesis - Homo sapiens (human) | Acetyl-CoA | C00024 | Glycolysis / Gluconeogenesis - Homo sapiens (human) |
| Glycolysis / Gluconeogenesis - Homo sapiens (human) | D-Glucose | C00031 | Glycolysis / Gluconeogenesis - Homo sapiens (human) |
| Glycolysis / Gluconeogenesis - Homo sapiens (human) | Acetate | C00033 | Glycolysis / Gluconeogenesis - Homo sapiens (human) |
| Glycolysis / Gluconeogenesis - Homo sapiens (human) | Oxaloacetate | C00036 | Glycolysis / Gluconeogenesis - Homo sapiens (human) |
2. Run MetaProViz Analysis
Currently, MetaProViz contains four different modules, which include different methods and can be used independently from each other or in combination (see introduction for more details). Here we will go trough each of those modules and apply them to the example data.
Pre-processing
MetaProViz includes a pre-processing module with the
function Preprocessing() that has multiple parameters to
perform customize data processing.Feature_Filtering applies the 80%-filtering rule on the
metabolite features either on the whole dataset (=“Standard”) (Bijlsma et al. 2006) or per condition
(=“Modified”) (Wei et al. 2018). This
means that metabolites are removed were more than 20% of the samples
(all or per condition) have no detection. In case of the CoRe
experiment, the blank samples are ignored during feature filtering,
since often metabolites are released from a cell and not naturally
present in the culture media leading to no detection in the blank. With
the parameter Feature_Filt_Value we enable the adaptation
of the stringency of the filtering based on the experimental context.
For instance, patient tumour samples can contain many unknown subgroups
due to gender, age, stage etc., which leads to a metabolite being
detected in only 50% (or even less) of the tumour samples, hence in this
context it could be considered to change the
Feature_Filt_Value from the default (=0.8). If
Feature_Filtering = "None", no feature filtering is
performed. In the context of Feature_Filtering it is also
noteworthy that the function Pool_Estimation() can be used
to estimate the quality of the metabolite detection and will return a
list of metabolites that are variable across the different pool
measurements (pool = mixture of all experimental samples measured
several times during the LC-MS run) . Variable metabolite in the pool
sample should be removed from the data.
The parameter TIC_Normalization refers to Total Ion Count
(TIC) normalisation, which is often used with LC-MS derived metabolomics
data. If TIC_Normalization = TRUE, each feature
(=metabolite) in a sample is divided by the sum of all intensity value
(= total number of ions) for the sample and finally multiplied by a
constant ( = the mean of all samples total number of ions). Noteworthy,
TIC normalisation should not be used with small number of features (=
metabolites), since TIC assumes that on “average” the ion count of each
sample is equal if there were no instrument batch effects (Wulff and Mitchell 2018).
The parameter MVI refers to Missing Value Imputation (MVI)
and if MVI = TRUE half minimum (HM) missing value
imputation is performed per feature (= per metabolite). Here it is
important to mention that HM has been shown to perform well for missing
vales that are missing not at random (MNAR) (Wei
et al. 2018).
Lastly, the function Preprocessing() performs outlier
detection and adds a column “Outliers” into the DF, which can be used to
remove outliers. The parameter HotellinsConfidence can be
used to choose the confidence interval that should be used for the
Hotellins T2 outlier test (Hotelling
1931).
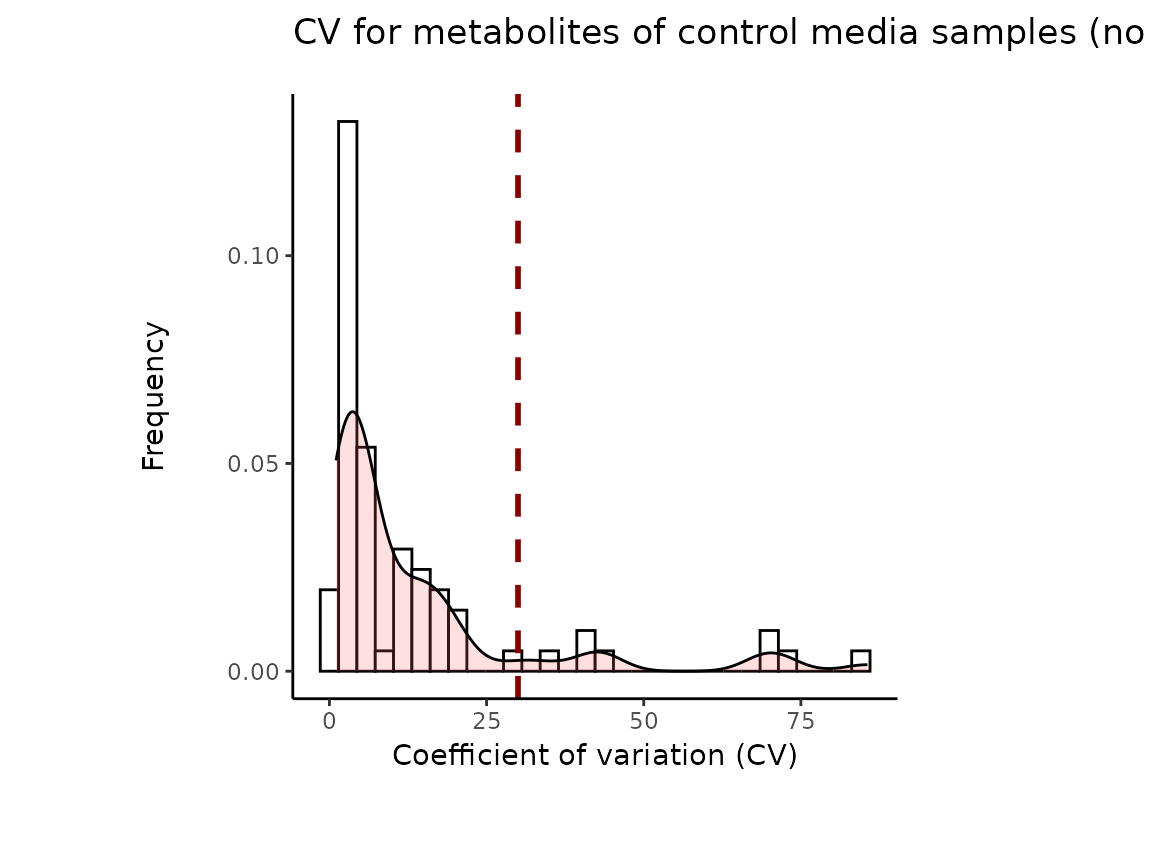
Since our example data contains pool samples, we will do
Pool_Estimation() before applying the
Preprocessing() function. This is important, since one
should remove the features (=metabolites) that are too variable prior to
performing any data transformations such as TIC as part of the
Preprocessing() function.
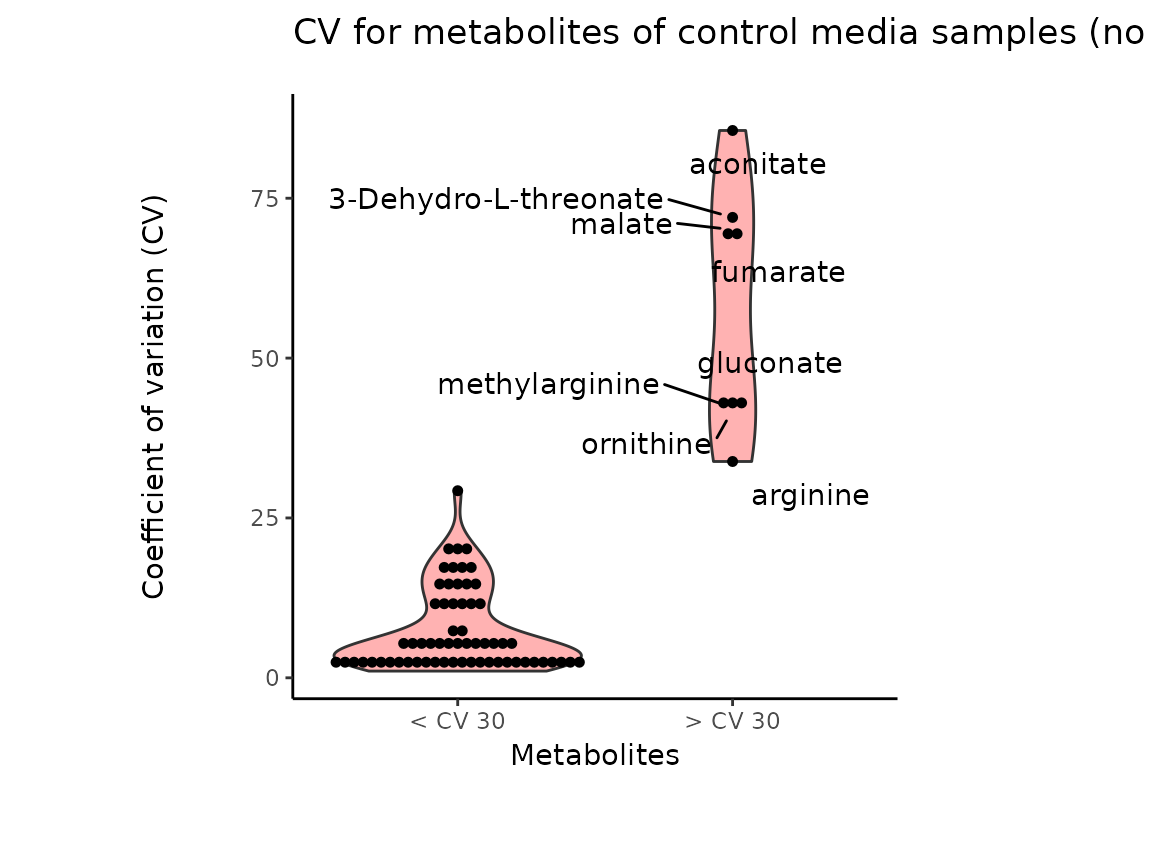
It is worth mentioning that the Coefficient of variation (CV) is
calculated by dividing the standard deviation (SD) by the mean. Hence CV
depends on the SD, which in turn works for normally distributed
data.
Pool_Estimation_result<- MetaProViz::PoolEstimation(InputData = Media[,-c(1:3)],
SettingsFile_Sample = Media[,1:3],
SettingsInfo = c(PoolSamples = "Pool", Conditions="Conditions"),
CutoffCV = 30)
Pool_Estimation_result_DF_CV <-Pool_Estimation_result[["DF"]][["CV"]]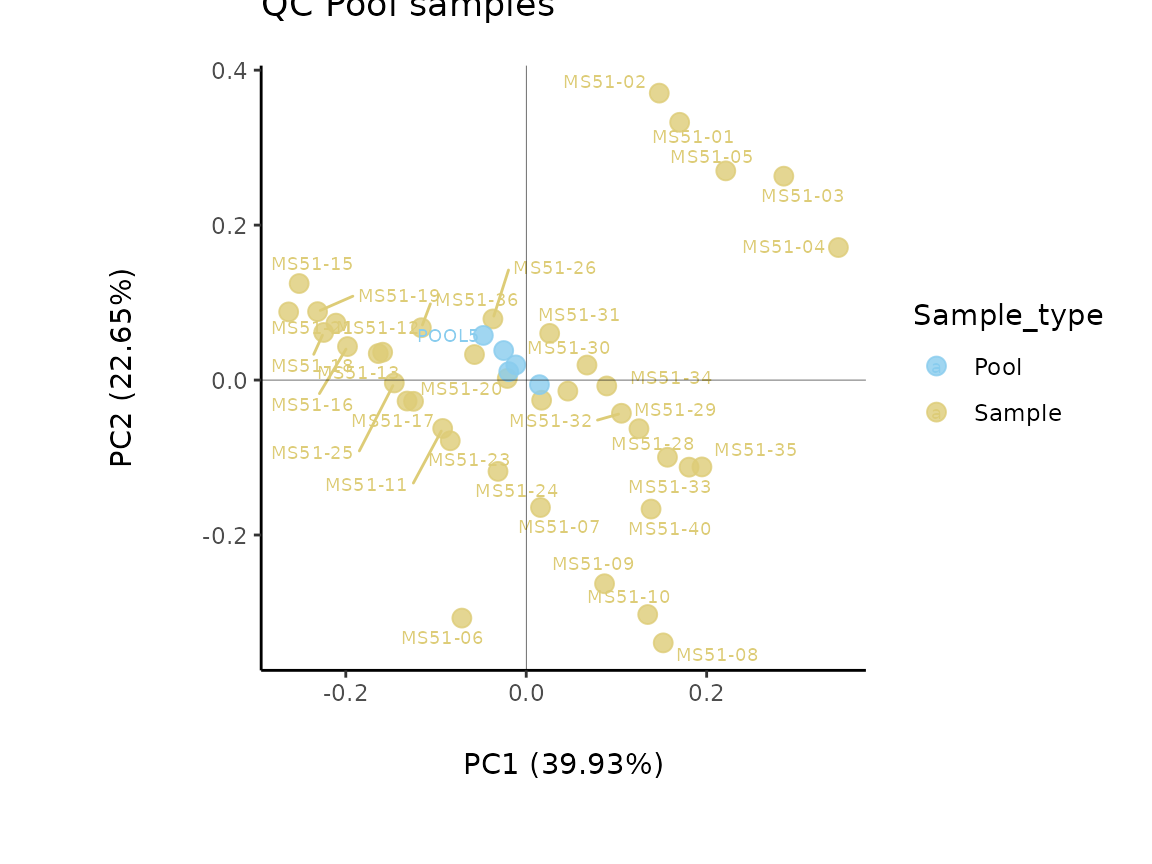

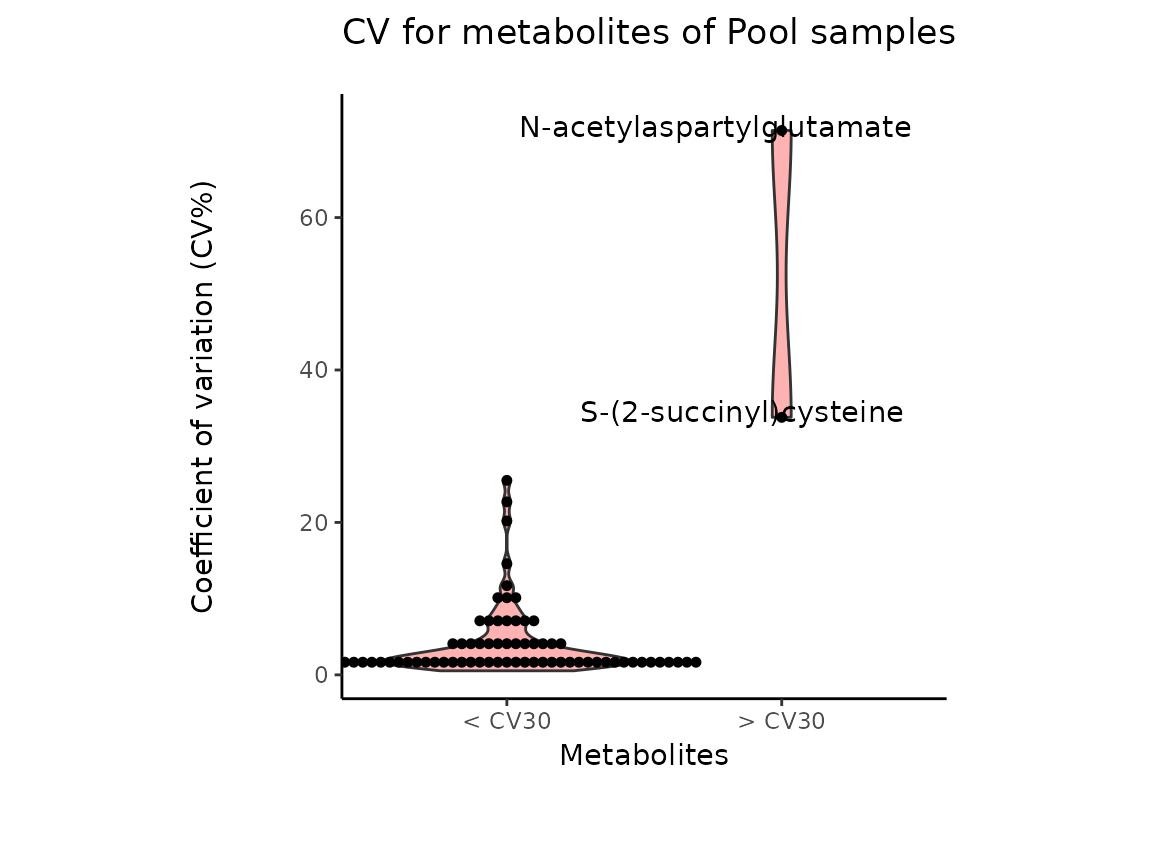
| Metabolite | CV | HighVar | MissingValuePercentage |
|---|---|---|---|
| valine-d8 | 1.744198 | FALSE | 0 |
| hipppuric acid-d5 | 1.253983 | FALSE | 0 |
| 2-hydroxyglutarate | 7.199141 | FALSE | 0 |
| 2-ketoglutarate | 2.766989 | FALSE | 0 |
| 3-Dehydro-L-threonate | 9.222780 | FALSE | 0 |
The results from the Pool_Estimation() is a table that has
the CVs. If there is a high variability, one should consider to remove
those features from the data. For the example data nothing needs to be
removed. If you have used internal standard in your experiment you
should specifically check their CV as this would indicate technical
issues (here valine-d8 and hippuric acid-d5).
Now we will apply the Preprocessing() function to the
example data and have a look at the output produced. You will notice
that all the chosen parameters and results are documented in messages.
All the results data tables, the Quality Control (QC) plots and outlier
detection plots are returned and can be easily viewed. Importantly, here
we are able to specify that we have a CoRe experiment setting the
parameter CoRe=TRUE, in which case a few additional data
processing steps are applied:1. Blank sample: This refers to media
samples where no cells have been cultured in, which will be used as
blank. In detail, the mean of the blank sample of a feature (=
metabolite) will be substracted from the values measured in each sample
for the same feature. In the column “Condition” of the
Experimental_design DF, you will need to label your blank samples with
“blank”.2. Growth factor or growth
rate: This refers to the different conditions and is either
based on cell count or protein quantification at the start of the
experiment (t0) and at the end of the experiment (t1) resulting in the
growth factor (t1/t0). Otherwise, one can experimentally estimate the
growth rate of each condition. Ultimately, this measure is used to
normalize the data, since the amount of growth will impact the
consumption and release of metabolites from the media and hence we need
to account for this. If you do not have this information, this will be
set to 1, yet be aware that this may affect the results.
You can pass these additional information via the parameter
Input_SettingsInfo, by passing the column name for the
CoRe_norm_factor in the Input_SettingsFile and
the condition name for the CoRe_media in the
Input_data file.
#Prepare the input:
Media_input <- Media%>%
subset(!Conditions=="Pool", select = -c(1:3))#remove pool samples and remove the information columns
Media_Metadata <- Media%>%
subset(!Conditions=="Pool", select = c(1:3))#remove pool samples and keep the information columns only
PreProcessing_res <- MetaProViz::PreProcessing(InputData=Media_input,
SettingsFile_Sample =Media_Metadata,
SettingsInfo = c(Conditions = "Conditions",
Biological_Replicates = "Biological_Replicates",
CoRe_norm_factor = "GrowthFactor",
CoRe_media = "blank"),
FeatureFilt = "Modified",
FeatureFilt_Value = 0.8,
TIC = TRUE,# As we have raw data we will perform total ion count norm
MVI=TRUE, #We assume the values are not missing at random and perform half minimum MVI
MVI_Percentage=50,
HotellinsConfidence = 0.99,# We perform outlier testing using 0.99 confidence interval
CoRe = TRUE)
# Now we can have a look at the results table:
Media_Preprocessed <- PreProcessing_res[["DF"]][["Preprocessing_output"]]#> For Consumption Release experiment we are using the method from Jain M. REF: Jain et. al, (2012), Science 336(6084):1040-4, doi: 10.1126/science.1218595.
#> FeatureFiltering: Here we apply the modified 80%-filtering rule that takes the class information (Column `Conditions`) into account, which additionally reduces the effect of missing values (REF: Yang et. al., (2015), doi: 10.3389/fmolb.2015.00004). Filtering value selected: 0.8
#> 3 metabolites where removed: N-acetylaspartylglutamate, hypotaurine, S-(2-succinyl)cysteine
#> Missing Value Imputation: Missing value imputation is performed, as a complementary approach to address the missing value problem, where the missing values are imputing using the `half minimum value`. REF: Wei et. al., (2018), Reports, 8, 663, doi:https://doi.org/10.1038/s41598-017-19120-0
#> NA values were found in Control_media samples for metabolites. For metabolites including NAs MVI is performed unless all samples of a metabolite are NA.
#> Metabolites with high NA load (>20%) in Control_media samples are: dihydroorotate.
#> Metabolites with only NAs (=100%) in Control_media samples are: hydroxyphenylpyruvate. Those NAs are set zero as we consider them true zeros
#> Total Ion Count (TIC) normalization: Total Ion Count (TIC) normalization is used to reduce the variation from non-biological sources, while maintaining the biological variation. REF: Wulff et. al., (2018), Advances in Bioscience and Biotechnology, 9, 339-351, doi:https://doi.org/10.4236/abb.2018.98022
#> 8 of variables have high variability (CV > 30) in the CoRe_media control samples. Consider checking the pooled samples to decide whether to remove these metabolites or not.
#> Warning in CoReNorm(InputData = TICRes, SettingsFile_Sample =
#> SettingsFile_Sample, : The CoRe_media samples MS51-06 were found to be
#> different from the rest. They will not be included in the sum of the CoRe_media
#> samples.
#> CoRe data are normalised by substracting mean (blank) from each sample and multiplying with the CoRe_norm_factor
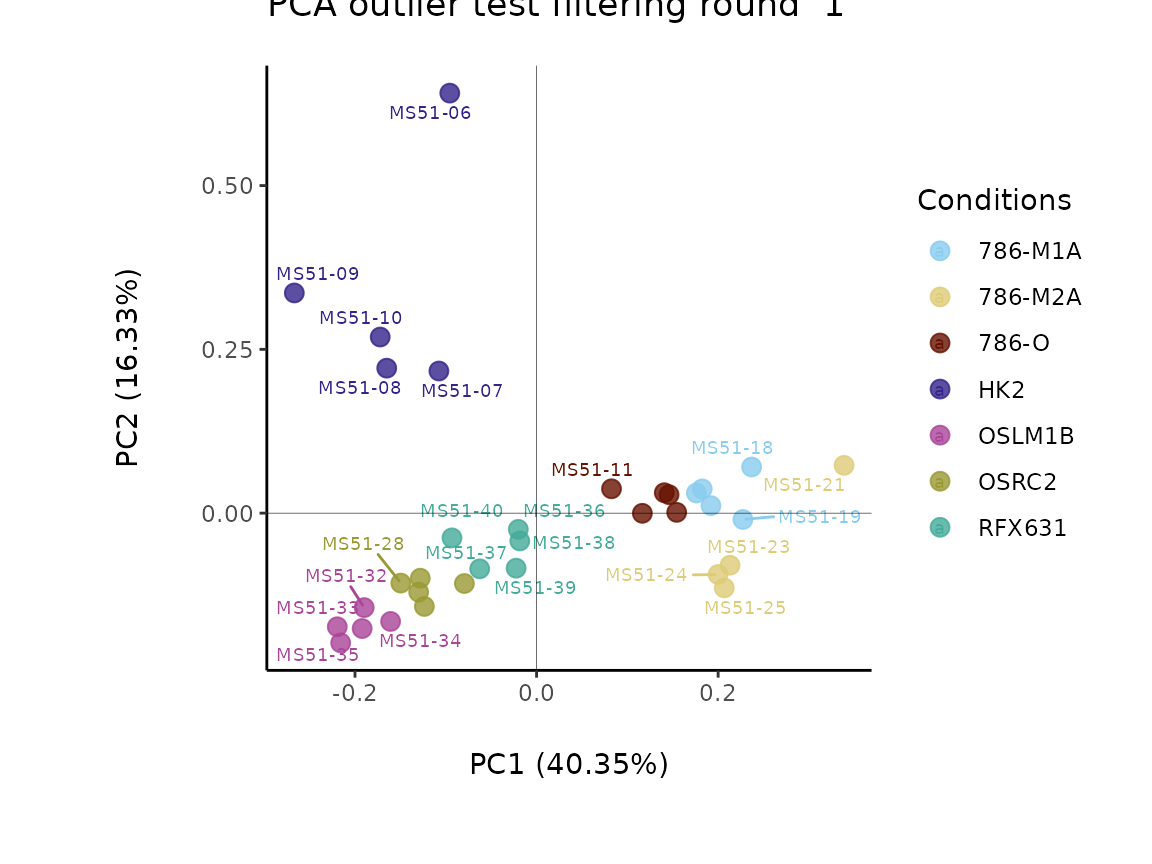
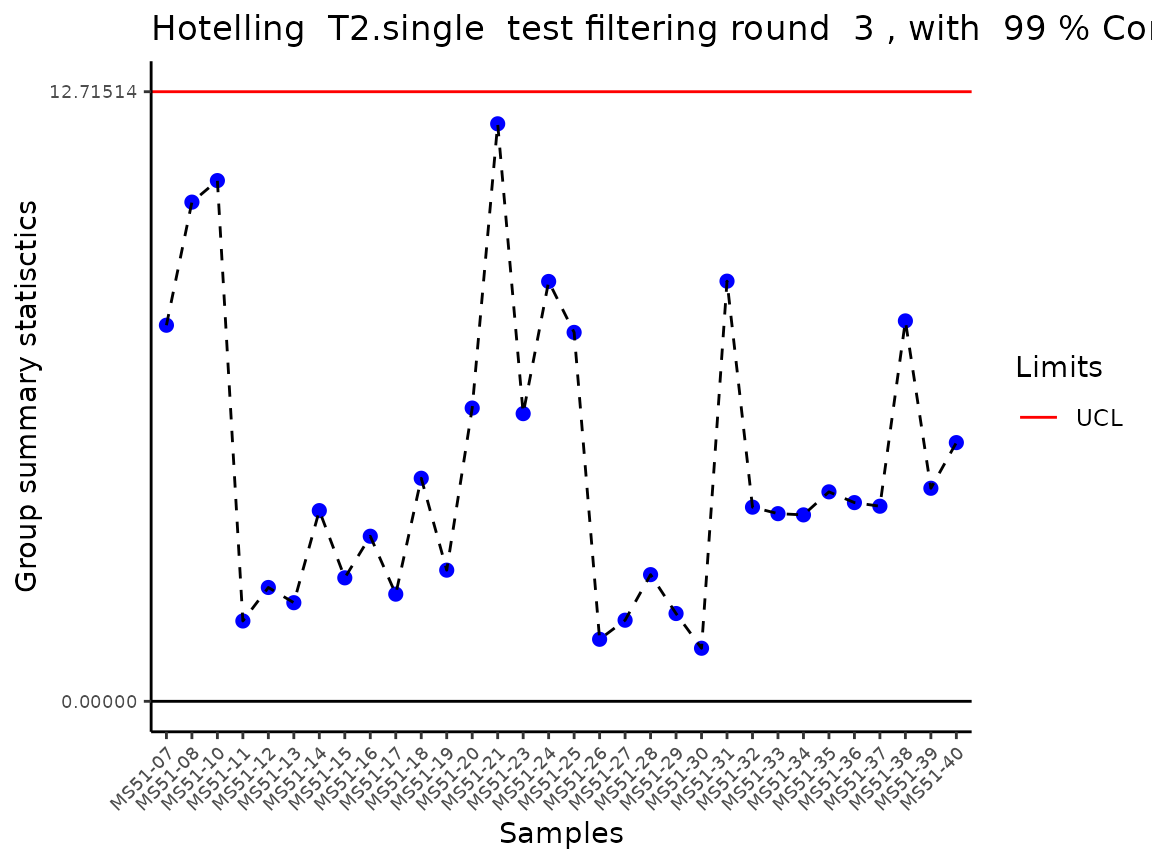
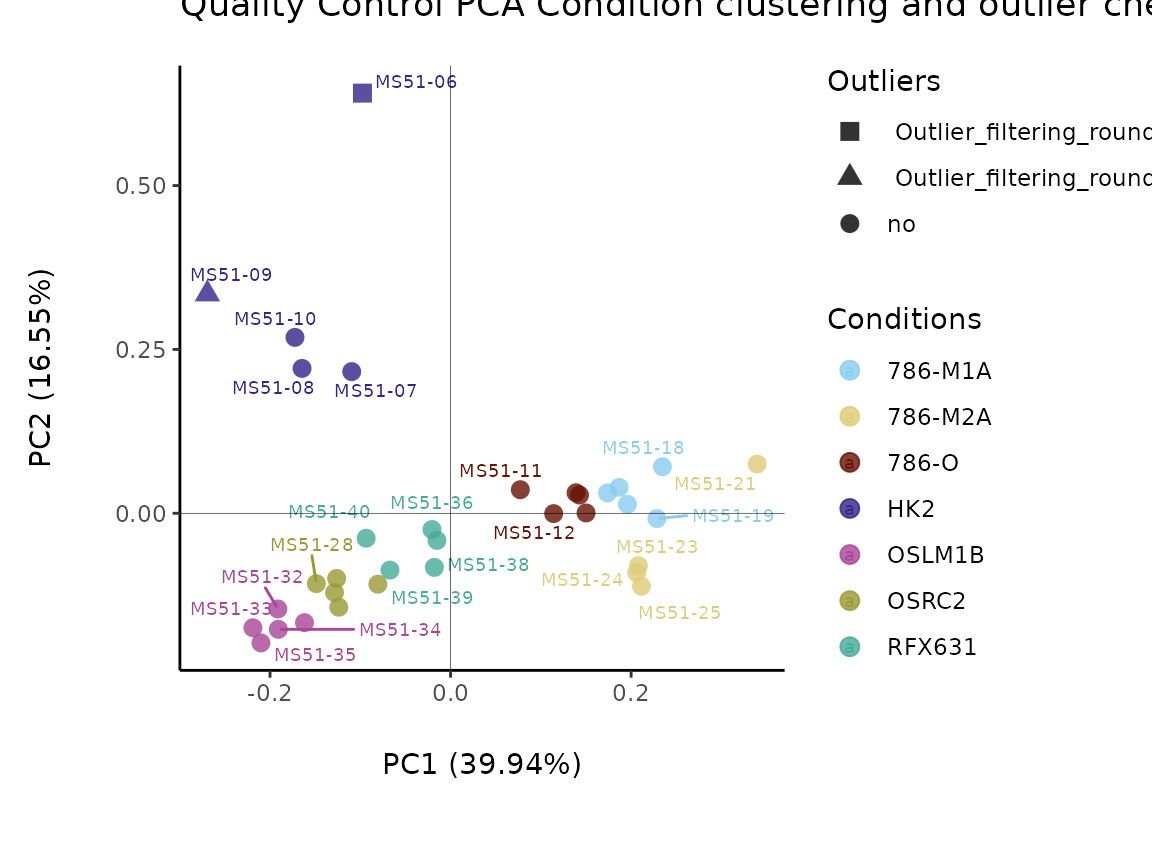
#> Outlier detection: Identification of outlier samples is performed using Hotellin's T2 test to define sample outliers in a mathematical way (Confidence = 0.99 ~ p.val < 0.01) (REF: Hotelling, H. (1931), Annals of Mathematical Statistics. 2 (3), 360–378, doi:https://doi.org/10.1214/aoms/1177732979). HotellinsConfidence value selected: 0.99
#> There are possible outlier samples in the data
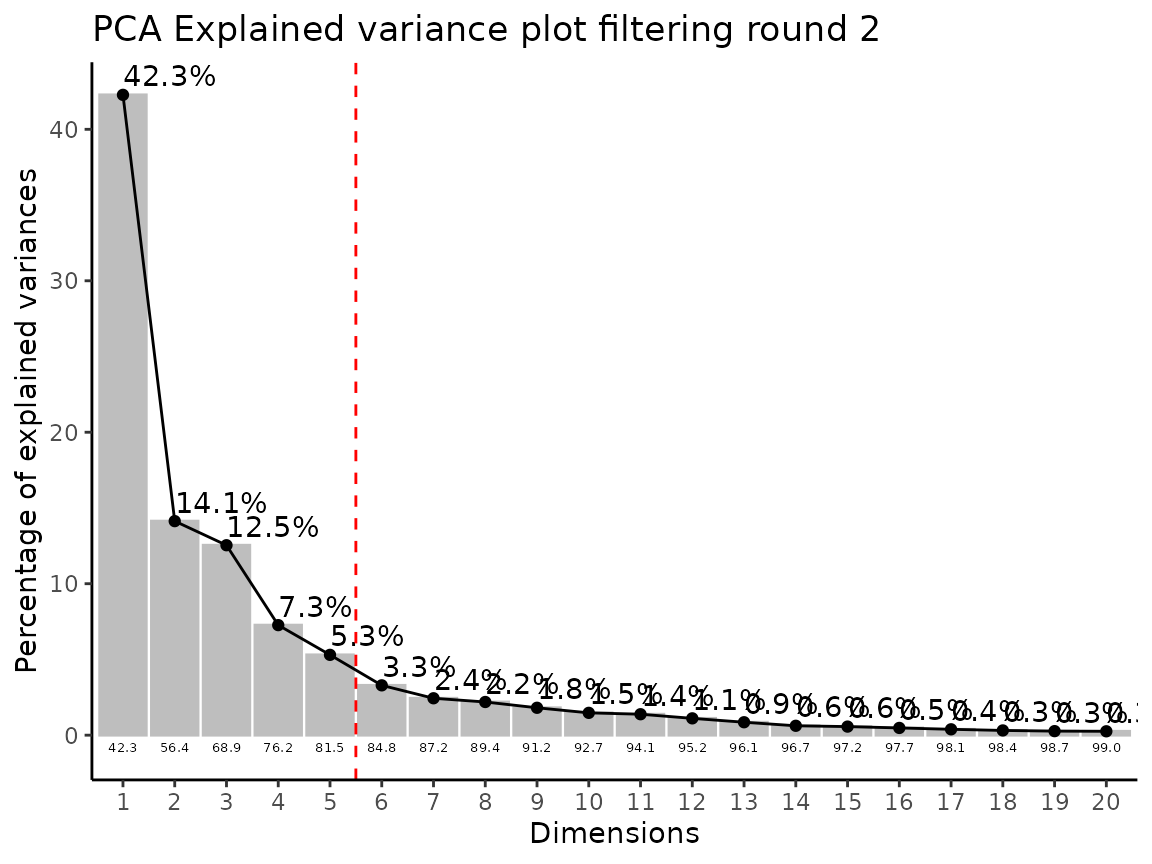
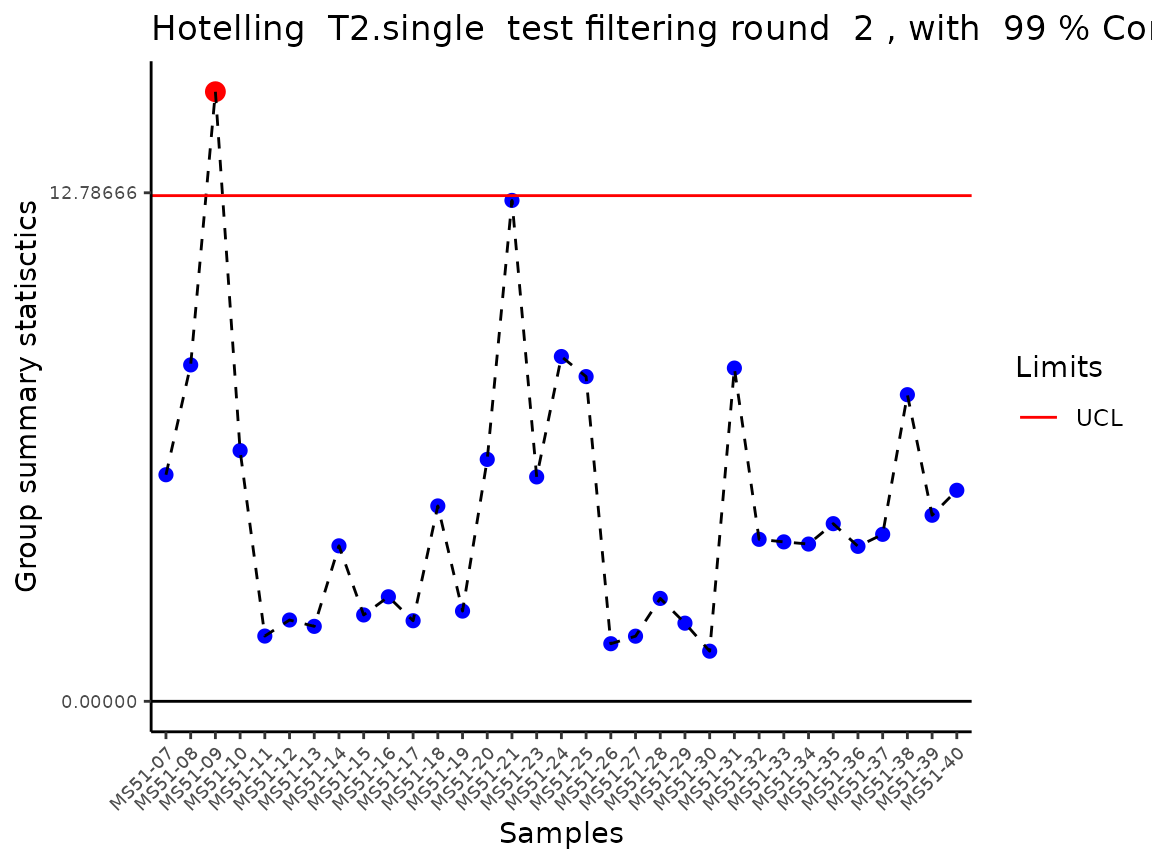
#> Filtering round 1 Outlier Samples: MS51-06
#> Filtering round 2 Outlier Samples: MS51-09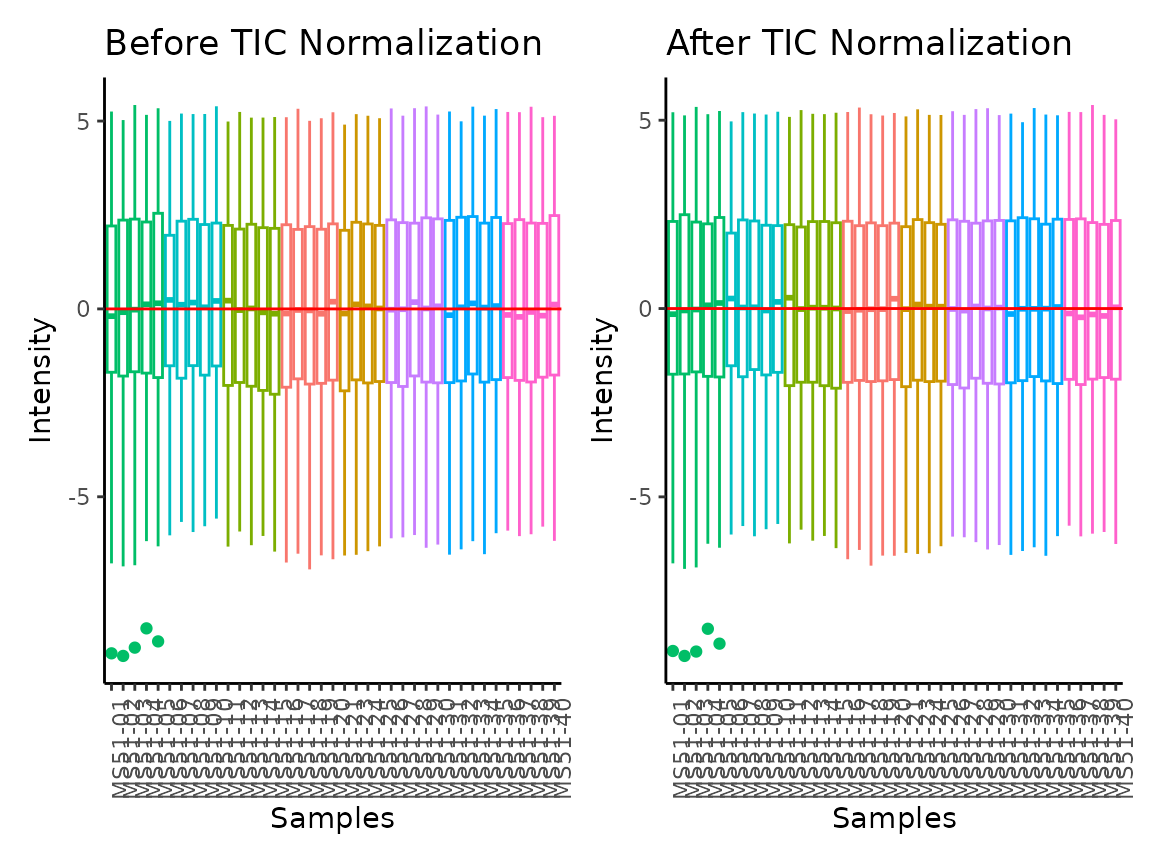





| Conditions | Biological_Replicates | GrowthFactor | Outliers | valine-d8 | hipppuric acid-d5 | 2-hydroxyglutarate | 2-ketoglutarate | 3-Dehydro-L-threonate | |
|---|---|---|---|---|---|---|---|---|---|
| MS51-06 | HK2 | 1 | 249.2817 | Outlier_filtering_round_1 | 6622682287 | 26658050746 | 5853665424 | 7527471565 | 318856944286 |
| MS51-07 | HK2 | 2 | 249.2817 | no | 5049281744 | 30218506574 | -868601575 | 1530912430 | 28673393701 |
| MS51-08 | HK2 | 3 | 249.2817 | no | -1441406385 | -11561761745 | 1108346025 | 3684365990 | 39313101752 |
| MS51-09 | HK2 | 4 | 249.2817 | Outlier_filtering_round_2 | 2563904070 | -10237061776 | -189633550 | -9097094735 | -67782920598 |
| MS51-10 | HK2 | 5 | 249.2817 | no | -6171779428 | -8419683053 | -50110899 | 3881816314 | -203574855 |
In the output table you can now see the column “Outliers” and for the
Condition HK2 CCM, we can see that based on Hotellin’s T2 test, samples
were detected as outliers in the first and second round of
filtering.
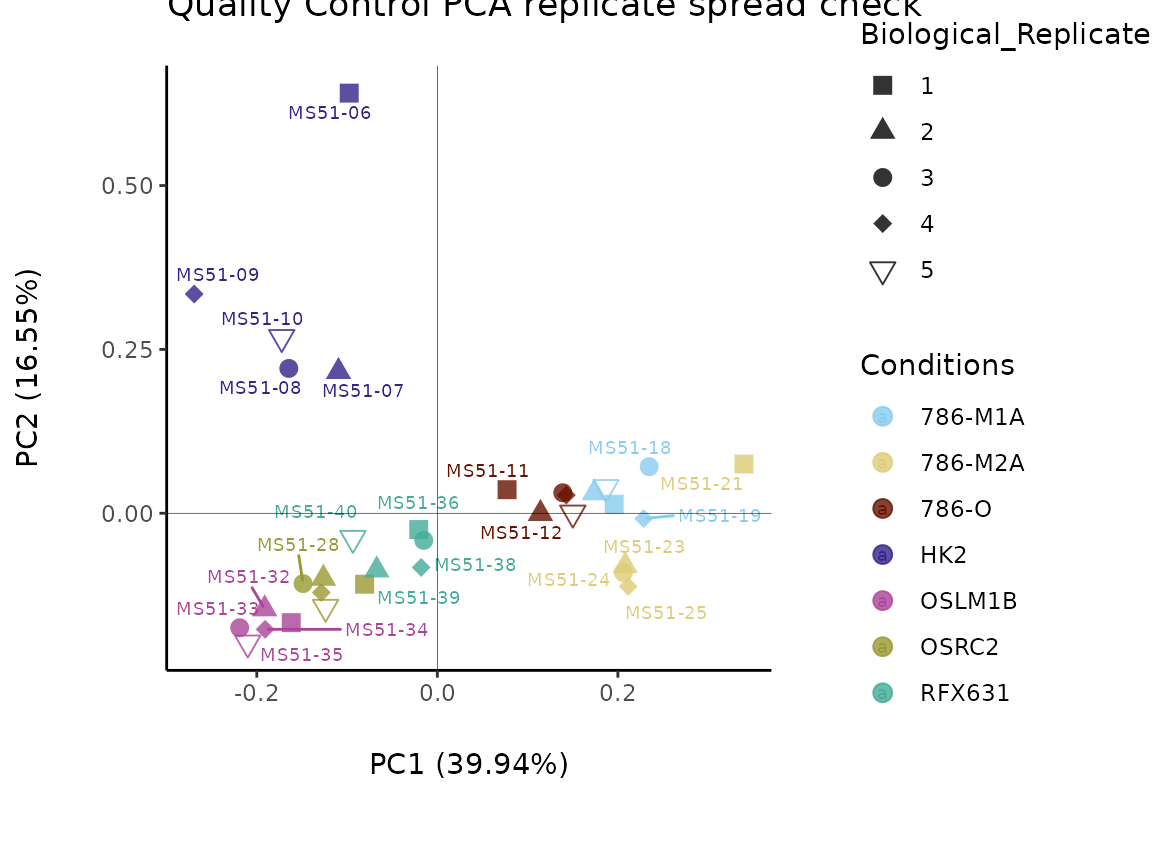
As part of the Preprocessing() function several plots are
generated and saved. Additionally, the ggplots are returned into the
list to enable further modifiaction using the ggplot syntax. These plots
include plots showing the outliers for each filtering round and other QC
plots.
As part of the MetaProViz visualization module one can
easily further customize the PCA plot and adapt color and shape for the
information of interest. You can see more below for the
VizPCA() function.
Before we proceed, we will remove the outlier:
In metabolomics, sometimes samples are injected (=measured) several
times, which can be termed as analytical replicates. The
MetaProViz pre-processing module includes the function
ReplicateSum(), which will summarize those and save the
results.
DMA
Differential Metabolite Analysis (DMA) between two
conditions (e.g. Tumour versus Healthy) usually calculates the Log2FC,
p-value, adjusted p-value and t-value. Yet, in a CoRe experiment the
normalized metabolite values can be either a negative value, if the
metabolite has been consumed from the media, or a positive value, if the
metabolite has been released from the cell into the culture media. Since
we can not calculate a Log2FC using negative values, we calculate the
absolute difference between the mean of Condition 1 versus the mean of
Condition 2. The absolute difference is log2 transformed in order to
make the values comparable between the different metabolites, resulting
in the Log2Dist. The result doesn’t consider whether one product is
larger than the other; it only looks at the magnitude of their
difference. To reflect the direction of change between the two
conditions we multiply with -1 if C1 < C2. By setting the paramteter
CoRe = TRUE, instead of calclulating the Log2FC, the Log2
Distance is calculated.
With the different parameters STAT_pval and
STAT_padj one can choose the statistical tests such as
t.test, wilcoxon test, limma, annova, kruskal walles, etc. (see function
reference for more information).
As input one can use the pre-processed data we have generated using the
Preprocessing module, but here one can of course use any DF
including metabolite values, even though we recommend to normalize the
data and remove outliers prior to DMA. Moreover, we require the
Input_SettingsFile_Sample including the sample metadata
with information which condition a sample corresponds to. Additionally,
we enable the user to provide a Plot_SettingsFile_Metab
containing the metadata for the features (metabolites), such as KEGG ID,
pathway, retention time, etc.
By defining the numerator and denominator as part of the
Input_SettingsInfo parameter, it is defined which
comparisons are performed:
1. one_vs_one (single comparison):
numerator=“Condition1”, denominator =“Condition2”
2. all_vs_one (multiple comparison): numerator=NULL,
denominator =“Condition”
3. all_vs_all (multiple comparison): numerator=NULL,
denominator =NULL (=default)
As input we will use the pre-processed data we have generated using the
Preprocessing module, but here one can of course use any DF
including metabolite values and information about the conditions that
should be compared (even though we recommend to normalize the data and
remove outliers prior to DMA).
In the example data we have seven different cell lines, healthy (HK2)
and cancer (ccRCC: 786-M1A, 786-M2A, 786-O, OSRC2, OSLM1B and RFX631)
and hence we can perform multiple different comparisons. The results can
be automatically saved and all the results are returned in a list with
the different data frames. If parameter Plot=TRUE, an overview Volcano
plot is generated and saved.
# Perform multiple comparison All_vs_One using annova:
DMA_Annova <- MetaProViz::DMA(InputData=Media_Preprocessed[,-c(1:6)],
SettingsFile_Sample=Media_Preprocessed[,c(1:4)],
SettingsInfo = c(Conditions="Conditions", Numerator=NULL, Denominator = "HK2"),
StatPval ="aov",
StatPadj="fdr",
SettingsFile_Metab = MappingInfo,
CoRe=TRUE)
#Inspect the DMA results tables:
DMA_786M1A_vs_HK2 <- DMA_Annova[["DMA"]][["786-M1A_vs_HK2"]]
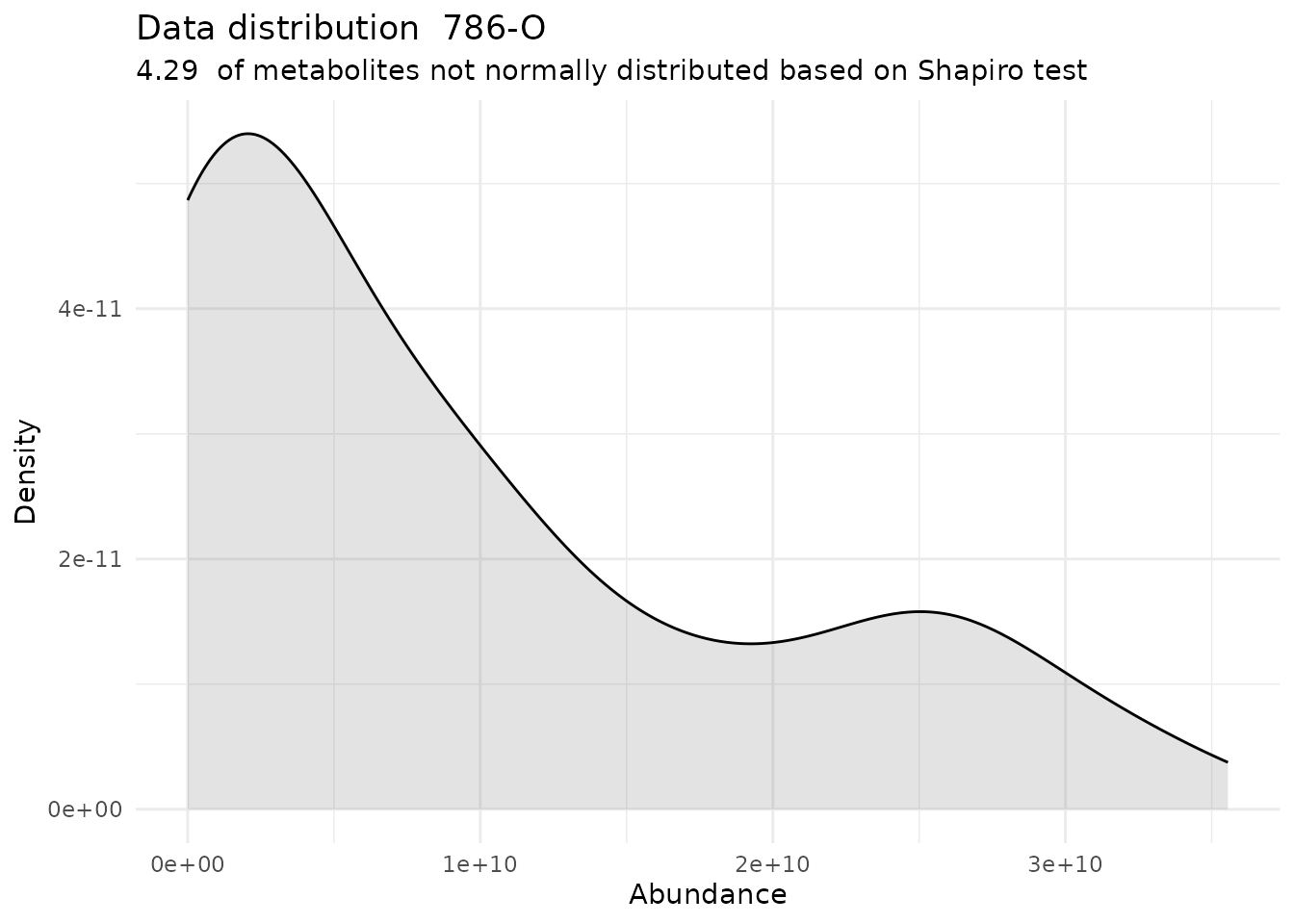
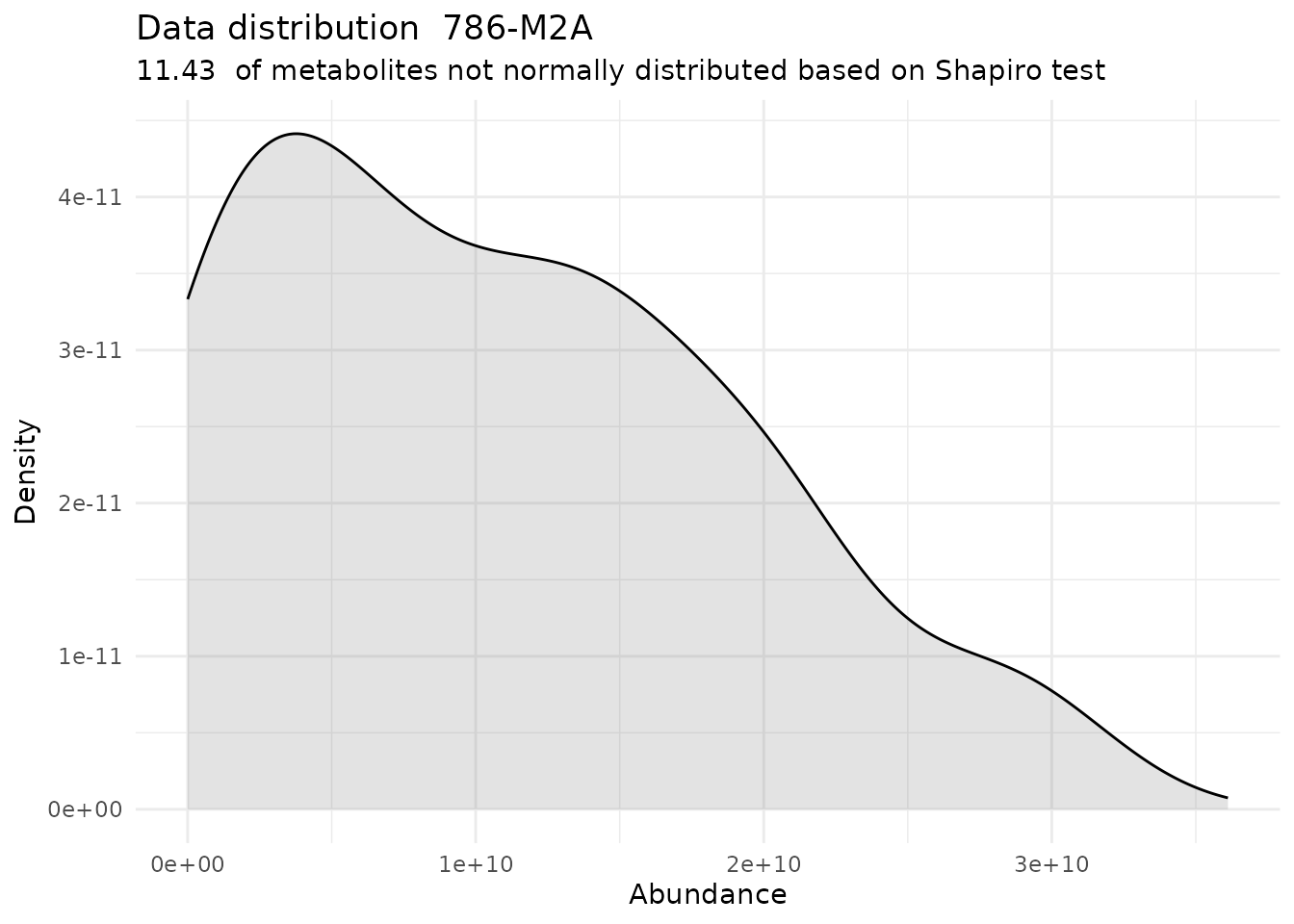
Shapiro <- DMA_Annova[["ShapiroTest"]][["DF"]][["Shapiro_result"]]#> There are no NA/0 values
#> For the condition HK2 82.35 % of the metabolites follow a normal distribution and 17.65 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition 786-O 95.71 % of the metabolites follow a normal distribution and 4.29 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition 786-M1A 97.14 % of the metabolites follow a normal distribution and 2.86 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition 786-M2A 88.57 % of the metabolites follow a normal distribution and 11.43 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition OSRC2 92.86 % of the metabolites follow a normal distribution and 7.14 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition OSLM1B 85.71 % of the metabolites follow a normal distribution and 14.29 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For the condition RFX631 97.14 % of the metabolites follow a normal distribution and 2.86 % of the metabolites are not-normally distributed according to the shapiro test. You have chosen aov, which is for parametric Hypothesis testing. `shapiro.test` ignores missing values in the calculation.
#> For 67.65% of metabolites the group variances are equal.
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> We added +1 to the mean value of metabolite(s) , since the mean of the replicate values where 0. This was not due to missing values (NA/0).
#> No condition was specified as numerator and HK2 was selected as a denominator. Performing multiple testing `all-vs-one` using aov.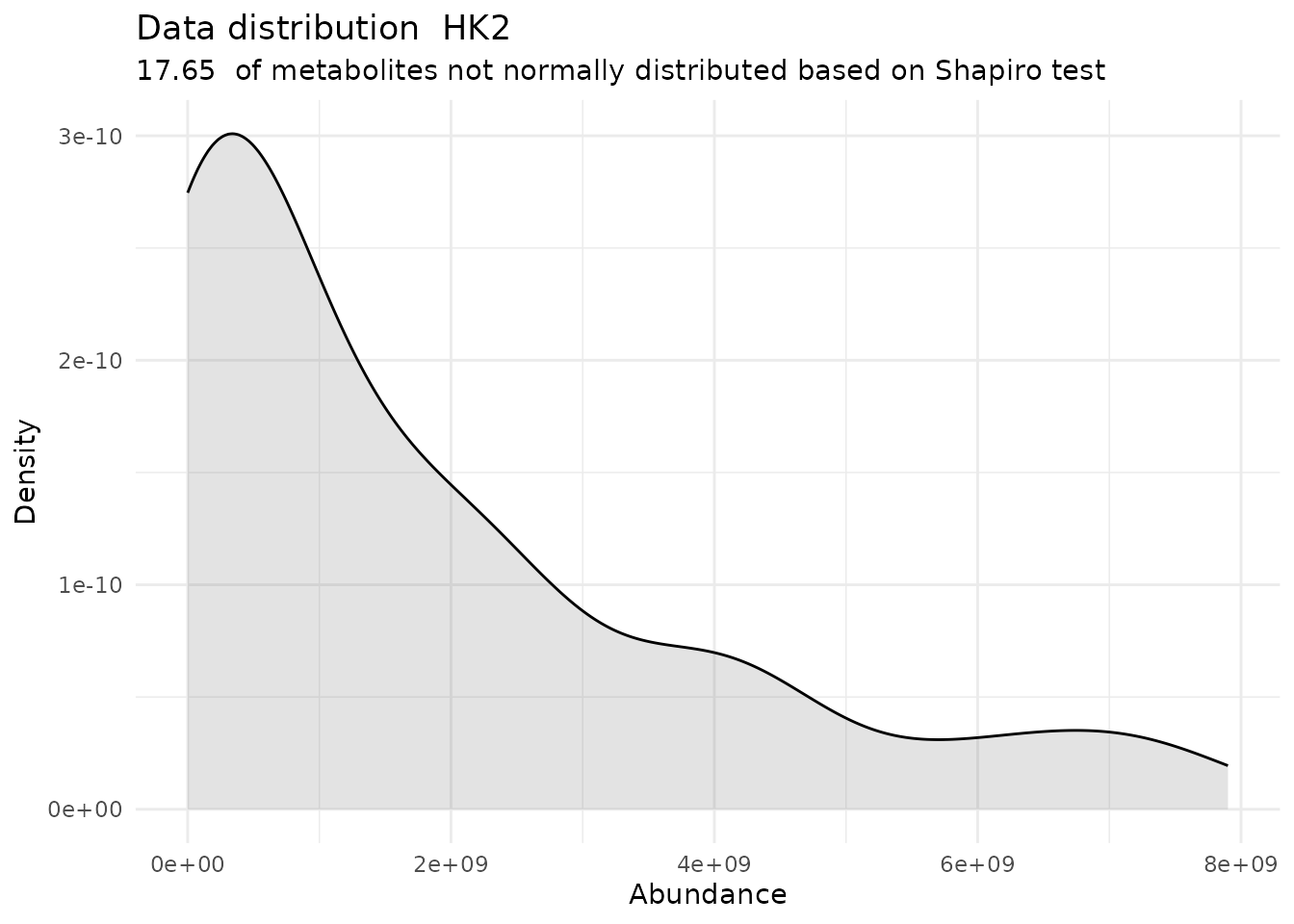






| Code | Metabolites with normal distribution [%] | Metabolites with not-normal distribution [%] | Shapiro p.val(2-hydroxyglutarate) | Shapiro p.val(2-ketoglutarate) |
|---|---|---|---|---|
| HK2 | 82.35 | 17.65 | 0.6833007 | 0.0446492 |
| 786-O | 95.71 | 4.29 | 0.4938675 | 0.3823712 |
| 786-M1A | 97.14 | 2.86 | 0.9979050 | 0.1384610 |
| 786-M2A | 88.57 | 11.43 | 0.5546558 | 0.5369470 |
| OSRC2 | 92.86 | 7.14 | 0.8899005 | 0.2007242 |
| OSLM1B | 85.71 | 14.29 | 0.4643014 | 0.9022803 |
| RFX631 | 97.14 | 2.86 | 0.9292099 | 0.0247568 |
| Metabolite | Log2(Distance) | p.adj | t.val | Mean_786-M1A | CoRe_786-M1A | Mean_HK2 | CoRe_HK2 | CoRe_specific | CoRe | MS51-16 | MS51-17 | MS51-18 | MS51-19 | MS51-20 | MS51-07 | MS51-08 | MS51-09 | MS51-10 | HMDB | KEGG.ID | KEGGCompound | Pathway |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aconitate | -32.58145 | 0.0000000 | 6426780519 | -6426780518.51619 | Consumed | -9.31322574615479e-07 | Consumed | Consumed | Consumed | -5865088200 | -6431671420 | -6503096254 | -6848155621 | -6485891098 | 1186119568 | -610503914 | -406215498 | -169400156 | HMDB0000072 | C00417 | cis-Aconitate | Citrate cycle (TCA cycle) |
| arginine | 36.63197 | 0.9411031 | -106493120308 | 106493120307.837 | Released | 5.7220458984375e-05 | Released | Released | Released | 216146922501 | 123584771692 | -184582255060 | 137947177034 | 239368985373 | 124885985748 | 174141953768 | -154239029796 | -144788909720 | HMDB0000517 | C00062 | L-Arginine | Amino acid metabolism |
| aspartate | -34.35422 | 0.0000000 | 21960976607 | -21960976606.516 | Consumed | -1.72853469848633e-06 | Consumed | Consumed | Consumed | -20078854995 | -23024873124 | -23695204780 | -22469858974 | -20536091160 | 1216173971 | 2386511960 | -2796694643 | -805991288 | HMDB0000191 | C00049 | L-Aspartate | Amino acid metabolism |
| betaine | 39.09360 | 0.0017858 | -586606639559 | 586606639558.529 | Released | 1.9073486328125e-06 | Released | Released | Released | 688358341457 | 490864850765 | 657311755851 | 724283773821 | 372214475898 | 222640096432 | -24221141664 | -466988404785 | 268569450018 | HMDB0000043 | C00719 | Betaine | Not assigned |
| carbamoyl phosphate | 29.63461 | 0.5228459 | -833502731 | 833502730.89944 | Released | 9.23871994018555e-07 | Released | Released | Released | 544741981 | 632898115 | 1989818101 | 816820279 | 183235178 | 200618839 | -354809141 | 549923002 | -395732700 | HMDB0001096 | C00169 | Carbamoyl phosphate | Purine metabolism |
Using the DMA results, we can now use the MetaProViz
visualization module and generate further customized Volcano plots
VizVolcano(). You can see some examples below.
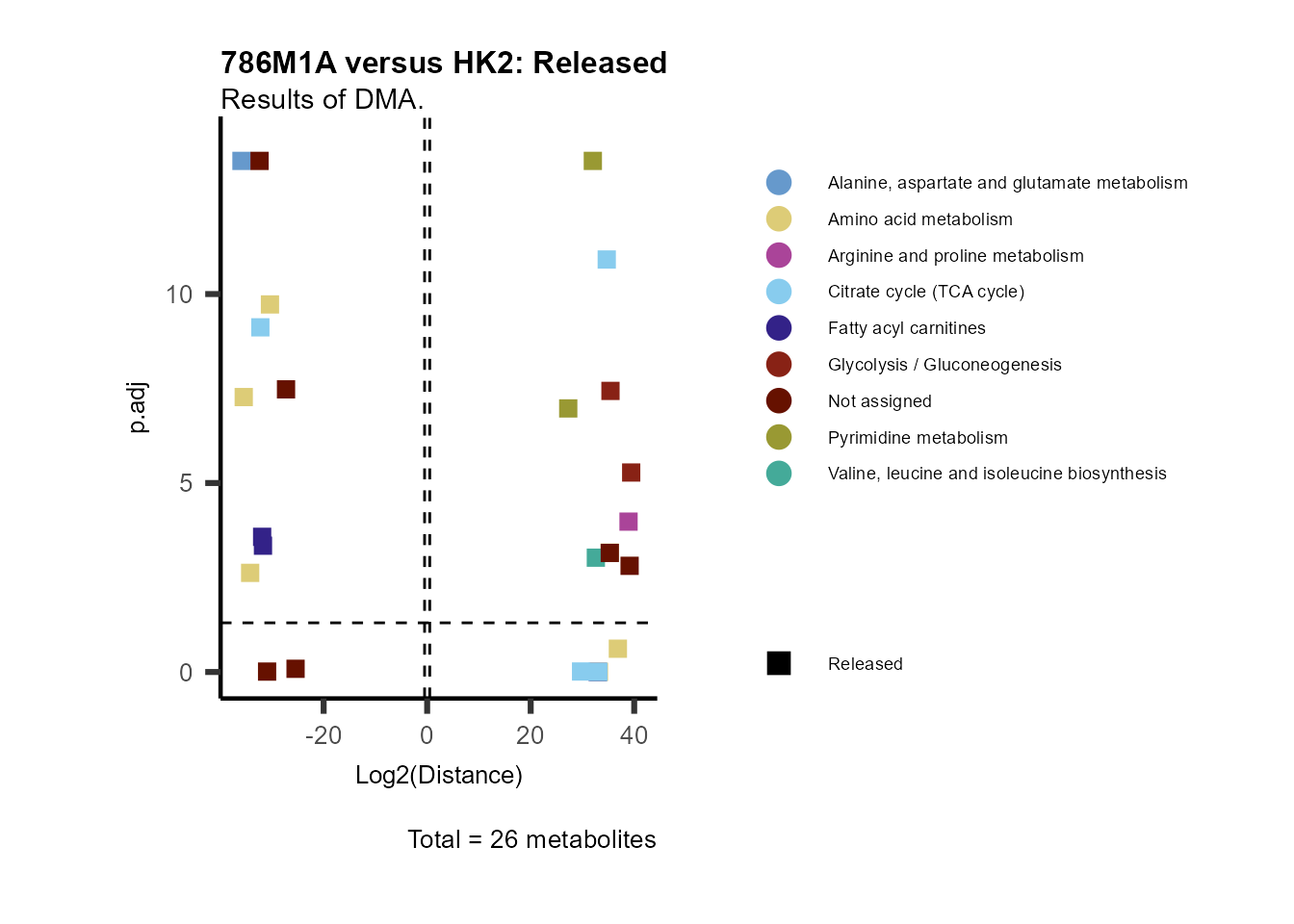
ORA using the DMA results
Over Representation Analysis (ORA) is a pathway enrichment analysis
(PEA) method that determines if a set of features (=metabolic pathways)
are over-represented in the selection of features (=metabolites) from
the data in comparison to all measured features (metabolites) using the
Fishers exact test. The selection of metabolites are usually the most
altered metabolites in the data, which can be selected by the top and
bottom t-values. Given that for CoRe data it is important to consider
weather a metabolite was consumed or released, it is sensible to perform
ORA on each metabolite cluster.
Of course, there are many other PEA methods such as the well known GSEA.
Here we do not aim to provide an extensive tool for different methods to
perform pathway enrichment analysis and only focus on ORA since we can
apply this to perform standard pathway enrichment as well as pathway
enrichment on clusters of metabolites. If you are interested in using
different pathway enrichment methods please check out specialized tools
such as decopupleR
(Badia-I-Mompel et al. 2022).
Here we will use the KEGG pathways (Kanehisa and
Goto 2000). Before we can perform ORA on the DMA results, we have
to ensure that the metabolite names match with the KEGG IDs or KEGG
trivial names. In general, the PathwayFile requirements are
column “term”, “Metabolite” and “Description”, and the
Input_data requirements are column “t.val” and column
“Metabolite”.
#Since we have performed multiple comparisons (all_vs_HK2), we will run ORA for each of this comparison
DM_ORA_res<- list()
comparisons <- names(DMA_Annova[["DMA"]])
for(comparison in comparisons){
#Ensure that the Metabolite names match with KEGG IDs or KEGG trivial names.
DMA <- DMA_Annova[["DMA"]][[comparison]]
DMA <- DMA[complete.cases(DMA),-1]%>%#we remove metabolites that do not have a KEGG ID/KEGG pathway
tibble::remove_rownames()%>%
column_to_rownames("KEGGCompound")#We use the KEGG trivial names to match with the KEGG pathways
#Perform ORA: Here we use
DM_ORA_res[[comparison]] <- MetaProViz::ClusterORA(InputData=DMA,
SettingsInfo=c(ClusterColumn="CoRe_specific", PathwayTerm= "term", PathwayFeature= "Metabolite"),
RemoveBackground=FALSE,#we do not have any background
PathwayFile=KEGG_Pathways,
PathwayName="KEGG",
minGSSize=3,
maxGSSize=1000)
}
#> Number of metabolites in cluster `Released in 786-M1A and Consumed HK2`: 10
#>
#> Number of metabolites in cluster `Consumed in 786-M1A and Released HK2`: 24
#> Number of metabolites in cluster `Consumed`: 14
#> Number of metabolites in cluster `Released`: 10
#> Number of metabolites in cluster `No Change`: 1
#> Number of metabolites in cluster `Released in 786-M2A and Consumed HK2`: 10
#> Number of metabolites in cluster `Consumed in 786-M2A and Released HK2`: 26
#> Number of metabolites in cluster `Consumed`: 14
#> Number of metabolites in cluster `Released`: 8
#> Number of metabolites in cluster `No Change`: 1
#> Number of metabolites in cluster `Released in 786-O and Consumed HK2`: 12
#> Number of metabolites in cluster `Consumed in 786-O and Released HK2`: 25
#> Number of metabolites in cluster `Consumed`: 12
#> Number of metabolites in cluster `Released`: 9
#> Number of metabolites in cluster `No Change`: 1
#> Number of metabolites in cluster `Released in OSLM1B and Consumed HK2`: 12
#> Number of metabolites in cluster `Consumed`: 12
#> Number of metabolites in cluster `Consumed in OSLM1B and Released HK2`: 16
#> Number of metabolites in cluster `Released`: 18
#> Number of metabolites in cluster `No Change`: 1
#> Number of metabolites in cluster `Released in OSRC2 and Consumed HK2`: 11
#> Number of metabolites in cluster `Consumed`: 13
#> Number of metabolites in cluster `Consumed in OSRC2 and Released HK2`: 25
#> Number of metabolites in cluster `Released`: 9
#> Number of metabolites in cluster `No Change`: 1
#> Number of metabolites in cluster `Released in RFX631 and Consumed HK2`: 6
#> Number of metabolites in cluster `Consumed`: 18
#> Number of metabolites in cluster `Released`: 10
#> Number of metabolites in cluster `Consumed in RFX631 and Released HK2`: 24
#> Number of metabolites in cluster `No Change`: 1
#Lets check how the results look like:
MC_ORA_786M1A_vs_HK2_Consumed <- DM_ORA_res[["786-M1A_vs_HK2"]][["DF"]][["Consumed"]]| GeneRatio | BgRatio | pvalue | p.adjust | qvalue | Metabolites_in_pathway | Count | Metabolites_in_Pathway | Percentage_of_Pathway_detected |
|---|---|---|---|---|---|---|---|---|
| 3/13 | 3/52 | 0.0129412 | 0.2394118 | 0.2315789 | L-Aspartate/L-Histidine/Pantothenate | 3 | 32 | 9.38 |
| 3/13 | 3/52 | 0.0129412 | 0.2394118 | 0.2315789 | (9Z)-Octadecenoic acid/Hexadecanoic acid/Octadecanoic acid | 3 | 58 | 5.17 |
| 3/13 | 4/52 | 0.0438415 | 0.5407123 | 0.5230218 | (9Z)-Octadecenoic acid/Hexadecanoic acid/Octadecanoic acid | 3 | 74 | 4.05 |
| 2/13 | 4/52 | 0.2573349 | 0.7934494 | 0.7674902 | Glycine/5-Oxoproline | 2 | 37 | 5.41 |
| 2/13 | 4/52 | 0.2573349 | 0.7934494 | 0.7674902 | L-Aspartate/L-Histidine | 2 | 47 | 4.26 |
MCA
Metabolite Clustering Analysis (MCA) is a module, which
includes different functions to enable clustering of metabolites into
groups based on logical regulatory rules. This can be particularly
useful if one has multiple conditions and aims to find patterns in the
data.
MCA_CoRe
This metabolite clustering method is based on logical regulatory
rules to sort metabolites into metabolite clusters. Here you
additionally need intracellular samples corresponding to the CoRe
samples.
Here we will define if a feature (= metabolite) is assigned into:
1. “UP”, which means a metabolite is
significantly up-regulated in the underlying comparison.
2. “DOWN”, which means a metabolite is
significantly down-regulated in the underlying comparison.
3. “No Change”, which means a metabolite does
not change significantly in the underlying comparison and/or is not
defined as up-regulated/down-regulated based on the Log2FC threshold
chosen.
Therebye “No Change” is further subdivided into four states:
1. “Not Detected”, which means a metabolite is
not detected in the underlying comparison.
2. “Not Significant”, which means a metabolite
is not significant in the underlying comparison.
3. “Significant positive”, which means a
metabolite is significant in the underlying comparison and the
differential metabolite abundance is positive, yet does not meet the
threshold set for “UP” (e.g. Log2FC >1 = “UP” and we have a
significant Log2FC=0.8).
4. “Significant negative”, which means a
metabolite is significant in the underlying comparison and the
differential metabolite abundance is negative, yet does not meet the
threshold set for “DOWN”.
Lastly, we also take into account the CoRe direction, meaning if a
metabolite was:
1. “Released”, which means is released into
the media in both conditions of the underlying comparison.
2. “Consumed”, which means is consumed from
the media in both conditions of the underlying comparison.
3. “Released/Consumed”, which means is
consumed/released in one condition, whilst the opposite occurs in the
second condition of the underlying comparison.
4. “Not Detected”, which means a metabolite is
not detected in the underlying comparison.
This definition is done individually for each comparison and will impact
in which metabolite cluster a metabolite is sorted into.
Since we have two comparisons (Intracellular and CoRe), we can choose
between different Background settings, which defines which features will
be considered for the clusters (e.g. you could include only features (=
metabolites) that are detected in both comparisons, removing the rest of
the features).The background methods backgroundMethod are
the following from 1.1. - 1.4. from most
restrictive to least restrictive:
1.1. Intra&CoRe: Most stringend background
setting and will lead to a small number of metabolites.
1.2. CoRe: Focus is on the metabolite
abundance of the CoRe.
1.3. Intra: Focus is on the metabolite
abundance of intracellular.
1.4. Intra|CoRe: Least stringent background
method, since a metabolite will be included in the input if it has been
detected on one of the two conditions.
Lastly, we will get clusters of metabolites that are defined by the
metabolite change in the two conditions. For example, if Alanine is “UP”
based on the thresholds in both comparisons it will be sorted into the
cluster “Core_UP”. As there are three 6-state6-state4 transitions
between the comparisons, the flows are summarised into smaller amount of
metabolite clusters using different Regulation Groupings (RG): 1.
RG1_All
2. RG2_Significant taking into account genes that are significant (UP,
DOWN, significant positive, significant negative)
3. RG3_SignificantChange only takes into account genes that have
significant changes (UP, DOWN).
In order to define which group a metabolite is assigned to, we set two
different thresholds. For intracellular those are based on the
differential metabolite abundance (Log2FC) and the
significance (e.g. p.adj). For the CoRe data this is based
on the Log2 Distance and the significance
(e.g. p.adj). For Log2FC we recommend a threshold of 0.5 or
1, whilst for the Log2 Distance one should check the
distance ranges and base the threhold on this.
Regulatory rules:
#Example of all possible flows:
MCA_CORE <- MetaProViz::MCA_rules(Method="CoRe")| Intra | CoRe | Core_Direction | RG1_All | R2_Significant | RG3_Change |
|---|---|---|---|---|---|
| DOWN | DOWN | Released | Intra DOWN+ CoRe DOWN_Released | Both_DOWN (Released) | Both_DOWN (Released) |
| DOWN | Not Detected | Not Detected | Intra DOWN+ CoRe Not Detected | None | None |
| DOWN | Not Significant | Released | Intra DOWN+ CoRe Not Significant_Released | None | None |
| DOWN | Significant Negative | Released | Intra DOWN+ CoRe Significant Negative_Released | Both_DOWN (Released) | None |
| DOWN | Significant Positive | Released | Intra DOWN+ CoRe Significant Positive_Released | Opposite (Released UP) | None |
| DOWN | UP | Released | Intra DOWN+ CoRe UP_Released | Opposite (Released UP) | Opposite (Released UP) |
| UP | DOWN | Released | Intra UP+ CoRe DOWN_Released | Opposite (Released DOWN) | Opposite (Released DOWN) |
| UP | Not Detected | Not Detected | Intra UP+ CoRe Not Detected | None | None |
| UP | Not Significant | Released | Intra UP+ CoRe Not Significant_Released | None | None |
| UP | Significant Negative | Released | Intra UP+ CoRe Significant Negative_Released | Opposite (Released UP) | None |
| UP | Significant Positive | Released | Intra UP+ CoRe Significant Positive_Released | Both_UP (Released) | None |
| UP | UP | Released | Intra UP+ CoRe UP_Released | Both_UP (Released) | Both_UP (Released) |
| Not Detected | DOWN | Released | Intra Not Detected+ CoRe DOWN_Released | CoRe_DOWN (Released) | CoRe_DOWN (Released) |
| Not Detected | Not Detected | Not Detected | Intra Not Detected+ CoRe Not Detected | None | None |
| Not Detected | Not Significant | Released | Intra Not Detected+ CoRe Not Significant_Released | None | None |
| Not Detected | Significant Negative | Released | Intra Not Detected+ CoRe Significant Negative_Released | None | None |
| Not Detected | Significant Positive | Released | Intra Not Detected+ CoRe Significant Positive_Released | None | None |
| Not Detected | UP | Released | Intra Not Detected+ CoRe UP_Released | CoRe_UP (Released) | CoRe_UP (Released) |
| Significant Negative | DOWN | Released | Intra Significant Negative+ CoRe DOWN_Released | Both_DOWN (Released) | CoRe_DOWN (Released) |
| Significant Negative | Not Detected | Not Detected | Intra Significant Negative+ CoRe Not Detected | None | None |
| Significant Negative | Not Significant | Released | Intra Significant Negative+ CoRe Not Significant_Released | None | None |
| Significant Negative | Significant Negative | Released | Intra Significant Negative+ CoRe Significant Negative_Released | None | None |
| Significant Negative | Significant Positive | Released | Intra Significant Negative+ CoRe Significant Positive_Released | None | None |
| Significant Negative | UP | Released | Intra Significant Negative+ CoRe UP_Released | Opposite (Released UP) | CoRe_UP (Released) |
| Significant Positive | DOWN | Released | Intra Significant Positive+ CoRe DOWN_Released | Opposite (Released DOWN) | CoRe_DOWN (Released) |
| Significant Positive | Not Detected | Not Detected | Intra Significant Positive+ CoRe Not Detected | None | None |
| Significant Positive | Not Significant | Released | Intra Significant Positive+ CoRe Not Significant_Released | None | None |
| Significant Positive | Significant Negative | Released | Intra Significant Positive+ CoRe Significant Negative_Released | None | None |
| Significant Positive | Significant Positive | Released | Intra Significant Positive+ CoRe Significant Positive_Released | None | None |
| Significant Positive | UP | Released | Intra Significant Positive+ CoRe UP_Released | Both_UP (Released) | CoRe_UP (Released) |
| Not Significant | DOWN | Released | Intra Not Significant+ CoRe DOWN_Released | CoRe_DOWN (Released) | CoRe_DOWN (Released) |
| Not Significant | Not Detected | Not Detected | Intra Not Significant+ CoRe Not Detected | None | None |
| Not Significant | Not Significant | Released | Intra Not Significant+ CoRe Not Significant_Released | None | None |
| Not Significant | Significant Negative | Released | Intra Not Significant+ CoRe Significant Negative_Released | None | None |
| Not Significant | Significant Positive | Released | Intra Not Significant+ CoRe Significant Positive_Released | None | None |
| Not Significant | UP | Released | Intra Not Significant+ CoRe UP_Released | CoRe_UP (Released) | CoRe_UP (Released) |
| DOWN | DOWN | Consumed | Intra DOWN+ CoRe DOWN_Consumed | Both_DOWN (Consumed) | Both_DOWN (Consumed) |
| DOWN | Not Detected | Not Detected | Intra DOWN+ CoRe Not Detected | None | None |
| DOWN | Not Significant | Consumed | Intra DOWN+ CoRe Not Significant_Consumed | None | None |
| DOWN | Significant Negative | Consumed | Intra DOWN+ CoRe Significant Negative_Consumed | Both_DOWN (Consumed) | None |
| DOWN | Significant Positive | Consumed | Intra DOWN+ CoRe Significant Positive_Consumed | Opposite (Consumed UP) | None |
| DOWN | UP | Consumed | Intra DOWN+ CoRe UP_Consumed | Opposite (Consumed UP) | Opposite (Consumed UP) |
| UP | DOWN | Consumed | Intra UP+ CoRe DOWN_Consumed | Opposite (Consumed DOWN) | Opposite (Consumed DOWN) |
| UP | Not Detected | Not Detected | Intra UP+ CoRe Not Detected | None | None |
| UP | Not Significant | Consumed | Intra UP+ CoRe Not Significant_Consumed | None | None |
| UP | Significant Negative | Consumed | Intra UP+ CoRe Significant Negative_Consumed | Opposite (Consumed UP) | None |
| UP | Significant Positive | Consumed | Intra UP+ CoRe Significant Positive_Consumed | Both_UP (Consumed) | None |
| UP | UP | Consumed | Intra UP+ CoRe UP_Consumed | Both_UP (Consumed) | Both_UP (Consumed) |
| Not Detected | DOWN | Consumed | Intra Not Detected+ CoRe DOWN_Consumed | CoRe_DOWN (Consumed) | CoRe_DOWN (Consumed) |
| Not Detected | Not Detected | Not Detected | Intra Not Detected+ CoRe Not Detected | None | None |
| Not Detected | Not Significant | Consumed | Intra Not Detected+ CoRe Not Significant_Consumed | None | None |
| Not Detected | Significant Negative | Consumed | Intra Not Detected+ CoRe Significant Negative_Consumed | None | None |
| Not Detected | Significant Positive | Consumed | Intra Not Detected+ CoRe Significant Positive_Consumed | None | None |
| Not Detected | UP | Consumed | Intra Not Detected+ CoRe UP_Consumed | CoRe_UP (Consumed) | CoRe_UP (Consumed) |
| Significant Negative | DOWN | Consumed | Intra Significant Negative+ CoRe DOWN_Consumed | Both_DOWN (Consumed) | CoRe_DOWN (Consumed) |
| Significant Negative | Not Detected | Not Detected | Intra Significant Negative+ CoRe Not Detected | None | None |
| Significant Negative | Not Significant | Consumed | Intra Significant Negative+ CoRe Not Significant_Consumed | None | None |
| Significant Negative | Significant Negative | Consumed | Intra Significant Negative+ CoRe Significant Negative_Consumed | None | None |
| Significant Negative | Significant Positive | Consumed | Intra Significant Negative+ CoRe Significant Positive_Consumed | None | None |
| Significant Negative | UP | Consumed | Intra Significant Negative+ CoRe UP_Consumed | Opposite (Consumed UP) | CoRe_UP (Consumed) |
| Significant Positive | DOWN | Consumed | Intra Significant Positive+ CoRe DOWN_Consumed | Opposite (Consumed DOWN) | CoRe_DOWN (Consumed) |
| Significant Positive | Not Detected | Not Detected | Intra Significant Positive+ CoRe Not Detected | None | None |
| Significant Positive | Not Significant | Consumed | Intra Significant Positive+ CoRe Not Significant_Consumed | None | None |
| Significant Positive | Significant Negative | Consumed | Intra Significant Positive+ CoRe Significant Negative_Consumed | None | None |
| Significant Positive | Significant Positive | Consumed | Intra Significant Positive+ CoRe Significant Positive_Consumed | None | None |
| Significant Positive | UP | Consumed | Intra Significant Positive+ CoRe UP_Consumed | Both_UP (Consumed) | CoRe_UP (Consumed) |
| Not Significant | DOWN | Consumed | Intra Not Significant+ CoRe DOWN_Consumed | CoRe_DOWN (Consumed) | CoRe_DOWN (Consumed) |
| Not Significant | Not Detected | Not Detected | Intra Not Significant+ CoRe Not Detected | None | None |
| Not Significant | Not Significant | Consumed | Intra Not Significant+ CoRe Not Significant_Consumed | None | None |
| Not Significant | Significant Negative | Consumed | Intra Not Significant+ CoRe Significant Negative_Consumed | None | None |
| Not Significant | Significant Positive | Consumed | Intra Not Significant+ CoRe Significant Positive_Consumed | None | None |
| Not Significant | UP | Consumed | Intra Not Significant+ CoRe UP_Consumed | CoRe_UP (Consumed) | CoRe_UP (Consumed) |
| DOWN | DOWN | Released/Consumed | Intra DOWN + CoRe DOWN_Released/Consumed | Both_DOWN (Released/Consumed) | Both_DOWN (Released/Consumed) |
| DOWN | Not Detected | Not Detected | Intra DOWN + CoRe Not Detected | None | None |
| DOWN | Not Significant | Released/Consumed | Intra DOWN + CoRe Not Significant_Released/Consumed | None | None |
| DOWN | Significant Negative | Released/Consumed | Intra DOWN + CoRe Significant Negative_Released/Consumed | Both_DOWN (Released/Consumed) | None |
| DOWN | Significant Positive | Released/Consumed | Intra DOWN + CoRe Significant Positive_Released/Consumed | Opposite (Released/Consumed UP) | None |
| DOWN | UP | Released/Consumed | Intra DOWN + CoRe UP_Released/Consumed | Opposite (Released/Consumed UP) | Opposite (Released/Consumed UP) |
| UP | DOWN | Released/Consumed | Intra UP + CoRe DOWN_Released/Consumed | Opposite (Released/Consumed DOWN) | Opposite (Released/Consumed DOWN) |
| UP | Not Detected | Not Detected | Intra UP + CoRe Not Detected | None | None |
| UP | Not Significant | Released/Consumed | Intra UP + CoRe Not Significant_Released/Consumed | None | None |
| UP | Significant Negative | Released/Consumed | Intra UP + CoRe Significant Negative_Released/Consumed | Opposite (Released/Consumed UP) | None |
| UP | Significant Positive | Released/Consumed | Intra UP + CoRe Significant Positive_Released/Consumed | Both_UP (Released/Consumed) | None |
| UP | UP | Released/Consumed | Intra UP + CoRe UP_Released/Consumed | Both_UP (Released/Consumed) | Both_UP (Released/Consumed) |
| Not Detected | DOWN | Released/Consumed | Intra Not Detected + CoRe DOWN_Released/Consumed | CoRe_DOWN (Released/Consumed) | CoRe_DOWN (Released/Consumed) |
| Not Detected | Not Detected | Not Detected | Intra Not Detected + CoRe Not Detected | None | None |
| Not Detected | Not Significant | Released/Consumed | Intra Not Detected + CoRe Not Significant_Released/Consumed | None | None |
| Not Detected | Significant Negative | Released/Consumed | Intra Not Detected + CoRe Significant Negative_Released/Consumed | None | None |
| Not Detected | Significant Positive | Released/Consumed | Intra Not Detected + CoRe Significant Positive_Released/Consumed | None | None |
| Not Detected | UP | Released/Consumed | Intra Not Detected + CoRe UP_Released/Consumed | CoRe_UP (Released/Consumed) | CoRe_UP (Released/Consumed) |
| Significant Negative | DOWN | Released/Consumed | Intra Significant Negative + CoRe DOWN_Released/Consumed | Both_DOWN (Released/Consumed) | CoRe_DOWN (Released/Consumed) |
| Significant Negative | Not Detected | Not Detected | Intra Significant Negative + CoRe Not Detected | None | None |
| Significant Negative | Not Significant | Released/Consumed | Intra Significant Negative + CoRe Not Significant_Released/Consumed | None | None |
| Significant Negative | Significant Negative | Released/Consumed | Intra Significant Negative + CoRe Significant Negative_Released/Consumed | None | None |
| Significant Negative | Significant Positive | Released/Consumed | Intra Significant Negative + CoRe Significant Positive_Released/Consumed | None | None |
| Significant Negative | UP | Released/Consumed | Intra Significant Negative + CoRe UP_Released/Consumed | Opposite (Released/Consumed UP) | CoRe_UP (Released/Consumed) |
| Significant Positive | DOWN | Released/Consumed | Intra Significant Positive + CoRe DOWN_Released/Consumed | Opposite (Released/Consumed DOWN) | CoRe_DOWN (Released/Consumed) |
| Significant Positive | Not Detected | Not Detected | Intra Significant Positive + CoRe Not Detected | None | None |
| Significant Positive | Not Significant | Released/Consumed | Intra Significant Positive + CoRe Not Significant_Released/Consumed | None | None |
| Significant Positive | Significant Negative | Released/Consumed | Intra Significant Positive + CoRe Significant Negative_Released/Consumed | None | None |
| Significant Positive | Significant Positive | Released/Consumed | Intra Significant Positive + CoRe Significant Positive_Released/Consumed | None | None |
| Significant Positive | UP | Released/Consumed | Intra Significant Positive + CoRe UP_Released/Consumed | Both_UP (Released/Consumed) | CoRe_UP (Released/Consumed) |
| Not Significant | DOWN | Released/Consumed | Intra Not Significant + CoRe DOWN_Released/Consumed | CoRe_DOWN (Released/Consumed) | CoRe_DOWN (Released/Consumed) |
| Not Significant | Not Detected | Not Detected | Intra Not Significant + CoRe Not Detected | None | None |
| Not Significant | Not Significant | Released/Consumed | Intra Not Significant + CoRe Not Significant_Released/Consumed | None | None |
| Not Significant | Significant Negative | Released/Consumed | Intra Not Significant + CoRe Significant Negative_Released/Consumed | None | None |
| Not Significant | Significant Positive | Released/Consumed | Intra Not Significant + CoRe Significant Positive_Released/Consumed | None | None |
| Not Significant | UP | Released/Consumed | Intra Not Significant + CoRe UP_Released/Consumed | CoRe_UP (Released/Consumed) | CoRe_UP (Released/Consumed) |
Now we can load the corresponding pre-processed intracellular example
data for the comparison of 786M-1A versus HK2 (For the detailed
pre-processing please see the vignette “Standard Metabolomics”).
#Load the Pre-processed intracellular data:
Intra_DMA_786M1A_vs_HK2<- MetaProViz::ToyData(Data="IntraCells_DMA")
#Perform metabolite clustering:
MCA_CoRe_res <- MetaProViz::MCA_CoRe(InputData_Intra =Intra_DMA_786M1A_vs_HK2%>%rownames_to_column("Metabolite") ,
InputData_CoRe = DMA_786M1A_vs_HK2,
SettingsInfo_Intra=c(ValueCol="Log2FC",StatCol="p.adj", StatCutoff= 0.05, ValueCutoff=1),
SettingsInfo_CoRe=c(DirectionCol="CoRe", ValueCol="Log2(Distance)",StatCol="p.adj", StatCutoff= 0.05, ValueCutoff=28),
FeatureID= "Metabolite",
BackgroundMethod="Intra&CoRe",
FolderPath=NULL)
#Lets check how the results look like:
MCA_res <- MCA_CoRe_res[["MCA_CoRe_Results"]]
ClusterSummary <- MCA_CoRe_res[["MCA_CoRe_Summary"]]| Metabolite | Intra_DF_Cutoff | Intra_DF_Cutoff_Specific.x | CoRe_DF_Detected | CoRe_DF_Cutoff | CoRe_DF_Cutoff_Specific | BG_Method | RG1_All | RG2_Significant | RG3_Change |
|---|---|---|---|---|---|---|---|---|---|
| adenosine | No Change | Not Significant | FALSE | No Change | Not Detected | FALSE | Background = FALSE | Background = FALSE | Background = FALSE |
| ADP | No Change | Not Significant | FALSE | No Change | Not Detected | FALSE | Background = FALSE | Background = FALSE | Background = FALSE |
| betaine | No Change | Significant Negative | TRUE | UP | UP | TRUE | Intra Significant Negative + CoRe UP_Released | Opposite (Released UP) | CoRe_UP (Released) |
| creatine | No Change | Significant Positive | TRUE | UP | UP | TRUE | Intra Significant Positive + CoRe UP_Released/Consumed | Both_UP (Released/Consumed) | CoRe_UP (Released/Consumed) |
| Regulation Grouping | SiRCle cluster Name | Number of Features |
|---|---|---|
| RG2_Significant | Both_UP (Released/Consumed) | 2 |
| RG2_Significant | Both_DOWN (Released/Consumed) | 10 |
| RG2_Significant | Opposite (Consumed DOWN) | 2 |
| RG2_Significant | Opposite (Released UP) | 2 |
| RG2_Significant | Both_DOWN (Consumed) | 2 |
| RG2_Significant | CoRe_DOWN (Released/Consumed) | 4 |
| RG2_Significant | CoRe_UP (Released/Consumed) | 2 |
| RG2_Significant | CoRe_UP (Released) | 1 |
| RG2_Significant | CoRe_DOWN (Consumed) | 1 |
| RG3_Change | Background = FALSE | 151 |
| RG3_Change | None | 22 |
| RG3_Change | Both_UP (Released/Consumed) | 1 |
| RG3_Change | CoRe_DOWN (Released/Consumed) | 8 |
| RG3_Change | CoRe_DOWN (Consumed) | 5 |
| RG3_Change | Both_DOWN (Released/Consumed) | 6 |
| RG3_Change | Opposite (Released UP) | 1 |
| RG3_Change | CoRe_UP (Released) | 2 |
| RG3_Change | CoRe_UP (Released/Consumed) | 3 |
ORA on each metabolite cluster
As explained in detail above, Over Representation Analysis (ORA) is a pathway enrichment analysis (PEA) method. As ORA is based on the Fishers exact test it is perfect to test if a set of features (=metabolic pathways) are over-represented in the selection of features (= clusters of metabolites) from the data in comparison to all measured features (all metabolites). In detail,MC_ORA() will perform ORA on
each of the metabolite clusters using all metabolites as the background.
| HMDB | KEGG.ID | KEGGCompound | Pathway | |
|---|---|---|---|---|
| N-acetylaspartate | HMDB0000812 | C01042 | N-Acetyl-L-aspartate | Alanine, aspartate and glutamate metabolism |
| argininosuccinate | HMDB0000052 | C03406 | N-(L-Arginino)succinate | Alanine, aspartate and glutamate metabolism |
| N-acetylaspartylglutamate | HMDB0001067 | C12270 | N-Acetylaspartylglutamate | Alanine, aspartate and glutamate metabolism |
| tyrosine | HMDB0000158 | C00082 | L-Tyrosine | Amino acid metabolism |
| asparagine | HMDB0000168 | C00152 | L-Asparagine | Amino acid metabolism |
| glutamate | HMDB0000148 | C00025 | L-Glutamate | Amino acid metabolism |
MC_ORA_result<- MetaProViz::ClusterORA(InputData=MCA_CoRe_res[["MCA_CoRe_Results"]]%>%column_to_rownames("Metabolite"),
SettingsInfo=c(ClusterColumn="RG2_Significant",
BackgroundColumn="BG_Method",
PathwayTerm= "Pathway", #This is the column name including the pathways names
PathwayFeature= "Metabolite"),
RemoveBackground=TRUE,
PathwayFile=MappingInfo%>%rownames_to_column("Metabolite"),
PathwayName="KEGG",
minGSSize=3,
maxGSSize=1000 ,
SaveAs_Table= "csv")
#> Number of metabolites in cluster `None`: 22
#> Number of metabolites in cluster `Both_UP (Released/Consumed)`: 2
#> Number of metabolites in cluster `Both_DOWN (Released/Consumed)`: 10
#> Number of metabolites in cluster `Opposite (Consumed DOWN)`: 2
#> Number of metabolites in cluster `Opposite (Released UP)`: 2
#> Number of metabolites in cluster `Both_DOWN (Consumed)`: 2
#> Number of metabolites in cluster `CoRe_DOWN (Released/Consumed)`: 4
#> Number of metabolites in cluster `CoRe_UP (Released/Consumed)`: 2
#> Number of metabolites in cluster `CoRe_UP (Released)`: 1
#> Number of metabolites in cluster `CoRe_DOWN (Consumed)`: 1
#Lets check how the results look like:
Both_UP_Released <- MC_ORA_result[["DF"]][["Both_UP (Released)"]]
Here we see that the pathways have a low amount of genes included that
were also part of the cluster and the pathways are not significant. This
is due to multiple factors, first we only start with a small number of
metabolites with KEGG IDs and secondly we only included metabolites if
they where detected in both, intracellular and CoRe samples (parameter
backgroundMethod="Intra&CoRe"). Hence, by for example
setting parameter backgroundMethod="Intra|CoRe", we will
obtain larger metabolite clusters.
3. Run MetaProViz Visualisation
The big advantages of the MetaProViz visualization
module is its flexible and easy usage, which we will showcase below and
that the figures are saved in a publication ready style and format. For
instance, the x- and y-axis size will always be adjusted for the amount
of samples or features (=metabolites) plotted, or in the case of Volcano
plot and PCA plot the axis size is fixed and not affected by figure
legends or title. In this way, there is no need for many adjustments and
the figures can just be dropped into the presentation or paper and are
all in the same style.
All the VizPlotName() functions are constructed in the same
way. Indeed, with the parameter Plot_SettingsInfo the user
can pass a named vector with information about the metadata column that
should be used to customize the plot by colour, shape or creating
individual plots, which will all be showcased for the different plot
types. Via the parameter Plot_SettingsFile the user can
pass the metadata DF, which can be dependent on the plot type for the
samples and/or the features (=metabolites). In case of both the
parameter is named Plot_SettingsFile_Sample and
Plot_SettingsFile_Metab.
In each of those Plot_Settings, the user can label color and/or shape
based on additional information (e.g. Pathway information, Cluster
information or other other demographics like gender). Moreover, we also
enable to plot individual plots where applicable based on those MetaData
(e.g. one plot for each metabolic pathway).
For this we need a metadata table including information about our
samples that could be relevant to e.g. color code:
MetaData_Sample <- Media_Preprocessed[,c(1:2)]%>%
mutate(Status = case_when(Conditions=="HK2" ~ 'Healthy',
TRUE ~ 'Cancer'))| Conditions | Biological_Replicates | Status | |
|---|---|---|---|
| MS51-07 | HK2 | 2 | Healthy |
| MS51-08 | HK2 | 3 | Healthy |
| MS51-09 | HK2 | 4 | Healthy |
| MS51-10 | HK2 | 5 | Healthy |
| MS51-11 | 786-O | 1 | Cancer |
| MS51-12 | 786-O | 2 | Cancer |
| MS51-13 | 786-O | 3 | Cancer |
| MS51-14 | 786-O | 4 | Cancer |
| MS51-15 | 786-O | 5 | Cancer |
| MS51-16 | 786-M1A | 1 | Cancer |
| MS51-17 | 786-M1A | 2 | Cancer |
| MS51-18 | 786-M1A | 3 | Cancer |
| MS51-19 | 786-M1A | 4 | Cancer |
| MS51-20 | 786-M1A | 5 | Cancer |
| MS51-21 | 786-M2A | 1 | Cancer |
| MS51-23 | 786-M2A | 2 | Cancer |
| MS51-24 | 786-M2A | 3 | Cancer |
| MS51-25 | 786-M2A | 4 | Cancer |
| MS51-26 | OSRC2 | 1 | Cancer |
| MS51-27 | OSRC2 | 2 | Cancer |
| MS51-28 | OSRC2 | 3 | Cancer |
| MS51-29 | OSRC2 | 4 | Cancer |
| MS51-30 | OSRC2 | 5 | Cancer |
| MS51-31 | OSLM1B | 1 | Cancer |
| MS51-32 | OSLM1B | 2 | Cancer |
| MS51-33 | OSLM1B | 3 | Cancer |
| MS51-34 | OSLM1B | 4 | Cancer |
| MS51-35 | OSLM1B | 5 | Cancer |
| MS51-36 | RFX631 | 1 | Cancer |
| MS51-37 | RFX631 | 2 | Cancer |
| MS51-38 | RFX631 | 3 | Cancer |
| MS51-39 | RFX631 | 4 | Cancer |
| MS51-40 | RFX631 | 5 | Cancer |
Moreover, we can use MetaData for our features (=Metabolites), which we
loaded with the MappingInfo and we can also add the
information on which cluster a metabolite was assigned to in the
MetaProViz::MCA() analysis above:
MetaData_Metab <-MappingInfo| HMDB | KEGG.ID | KEGGCompound | Pathway |
|---|---|---|---|
| HMDB0001067 | C12270 | N-Acetylaspartylglutamate | Alanine, aspartate and glutamate metabolism |
| HMDB0000158 | C00082 | L-Tyrosine | Amino acid metabolism |
| HMDB0000148 | C00025 | L-Glutamate | Amino acid metabolism |
| HMDB0250980 | NA | NA | Amino acid metabolism |
| NA | NA | NA | Amino acid metabolism |
| HMDB0004207 | NA | NA | Amino acid metabolism |
Noteworthy, here we can also use the KEGG pathways we used for the
pathway analysis.
PCA plots
Principal component analysis (PCA) is a dimensionality reduction
method that reduces all the measured features (=metabolites) of one
sample into a few features in the different principal components,
whereby each principal component can explain a certain percentage of the
variance between the different samples. Hence, this enables
interpretation of sample clustering based on the measured features
(=metabolites).
As mentioned above, PCA plots can be quite useful for quality control,
but of course it offers us many more opportunities, which will be
showcased here.
As input, we need a DF that contains the samples as rownames and the
features (=metabolites) as column names:
Input_PCA <- Media_Preprocessed[,-c(1:4)] #remove columns that include Metadata such as cell type,...| valine-d8 | hipppuric acid-d5 | 2-hydroxyglutarate | 2-ketoglutarate | 3-Dehydro-L-threonate | acetylcarnitine | acetylcholine | |
|---|---|---|---|---|---|---|---|
| MS51-07 | 5049281744 | 30218506574 | -868601575 | 1530912430 | 28673393701 | -67547592848 | -496576017 |
| MS51-08 | -1441406385 | -11561761745 | 1108346025 | 3684365990 | 39313101752 | -234677799645 | -124297109 |
| MS51-09 | 2563904070 | -10237061776 | -189633550 | -9097094735 | -67782920598 | 218513442831 | 526295913 |
| MS51-10 | -6171779428 | -8419683053 | -50110899 | 3881816314 | -203574855 | 83711949661 | 94577213 |
| MS51-11 | 27378617230 | 123649127347 | 4311754839 | 23286535748 | -16721754062 | -434945308541 | -183109349 |
| MS51-12 | 31998154622 | 99965859879 | 808357705 | 19381509727 | -16349278172 | -583665678018 | -998475065 |
Now lets check out the standard plot:
MetaProViz::VizPCA(InputData=Input_PCA)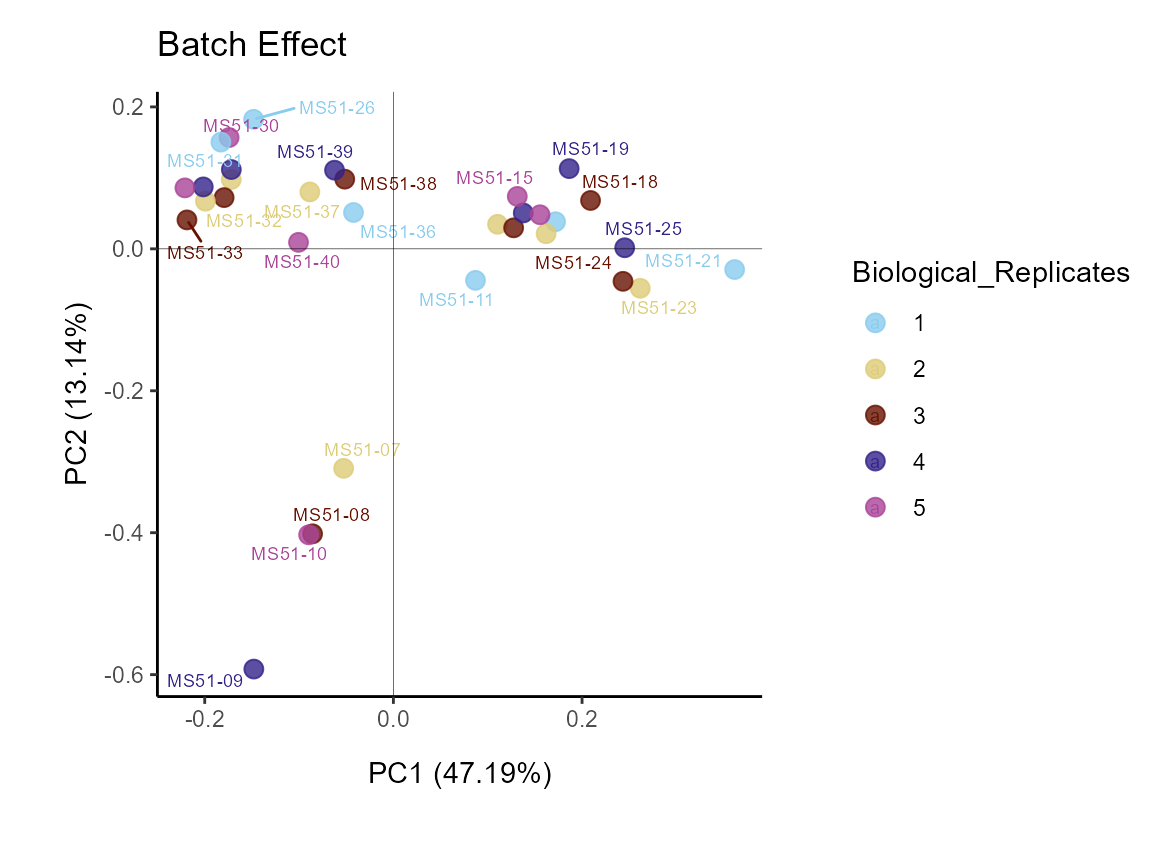
Figure: Standard Settings.
Next, we can interactively choose shape and color using the
additional information of interest from our Metadata. Especially for
complex data, such as patient data, it can be valuable to use different
demographics (e.g. age, gender, medication,…) for this. First lets check
if we have any batch effect by colour coding for the biological
replicates, which would be the case if the replicates cluster
together.
MetaProViz::VizPCA(SettingsInfo= c(color="Biological_Replicates"),
SettingsFile_Sample = MetaData_Sample ,
InputData=Input_PCA,
PlotName = "Batch Effect")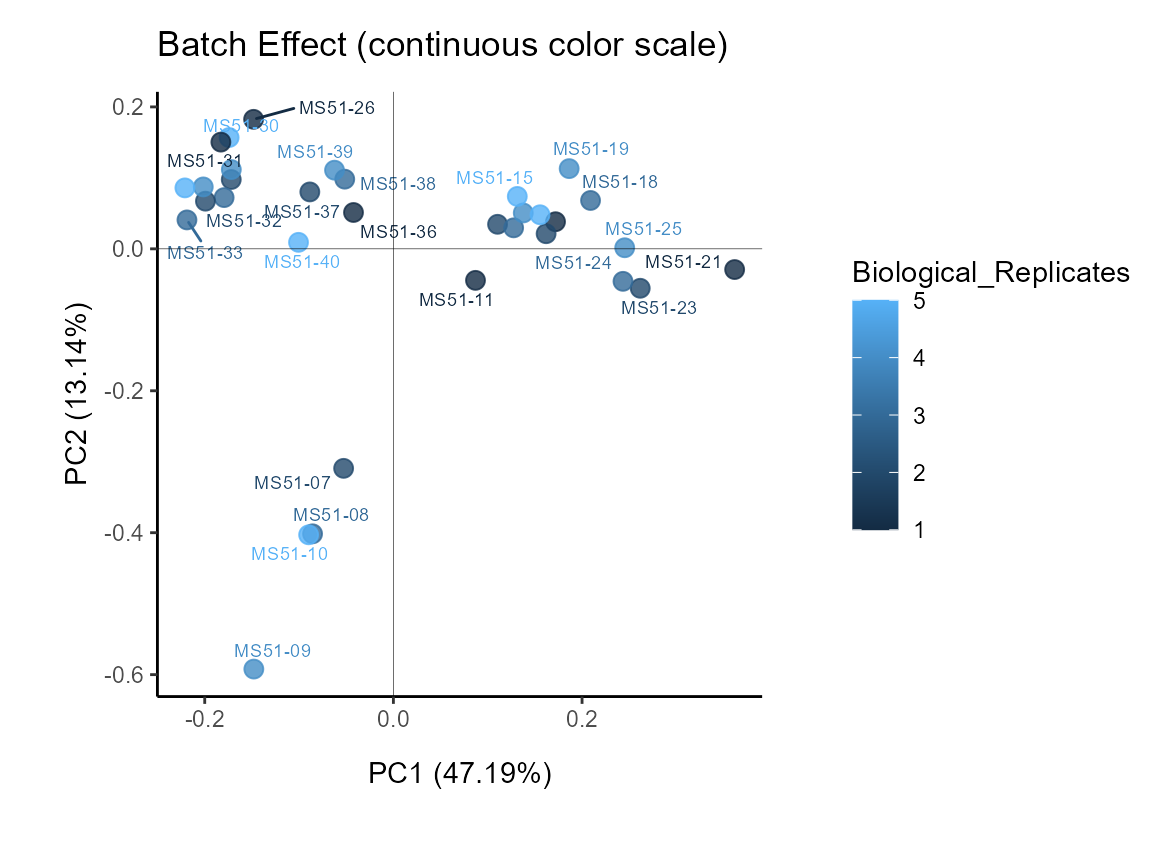
Figure: Do we have a batch effect?
Given the biological replicates are numeric, we can also set
color_scale to continuous:
MetaProViz::VizPCA(SettingsInfo= c(color="Biological_Replicates"),
SettingsFile_Sample = MetaData_Sample ,
InputData=Input_PCA,
ColorScale = "continuous",
PlotName = "Batch Effect (continuous color scale)")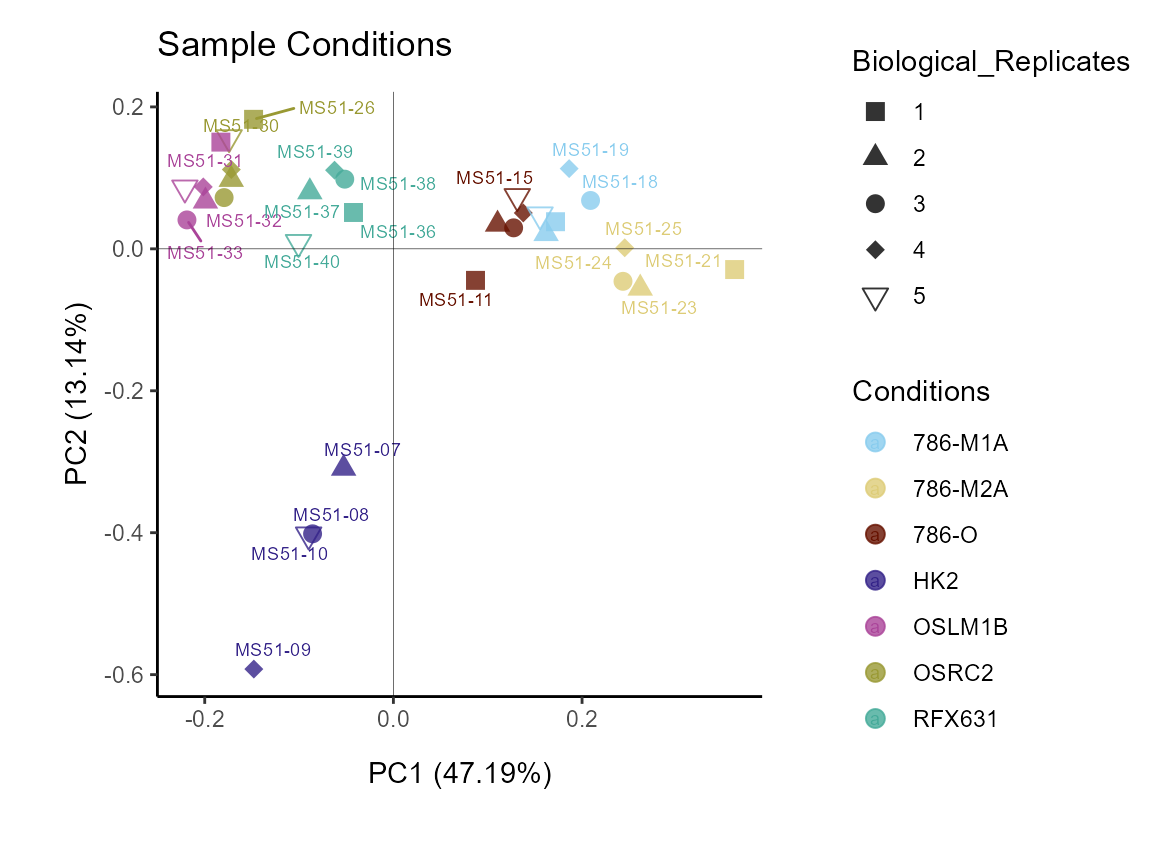
Figure: Do we have a batch effect?
Next, we can colour code for condition and use the biological
replicates in the shape parameter:
MetaProViz::VizPCA(SettingsInfo= c(color="Conditions", shape="Biological_Replicates"),
SettingsFile_Sample = MetaData_Sample ,
InputData=Input_PCA,
PlotName = "Sample Conditions")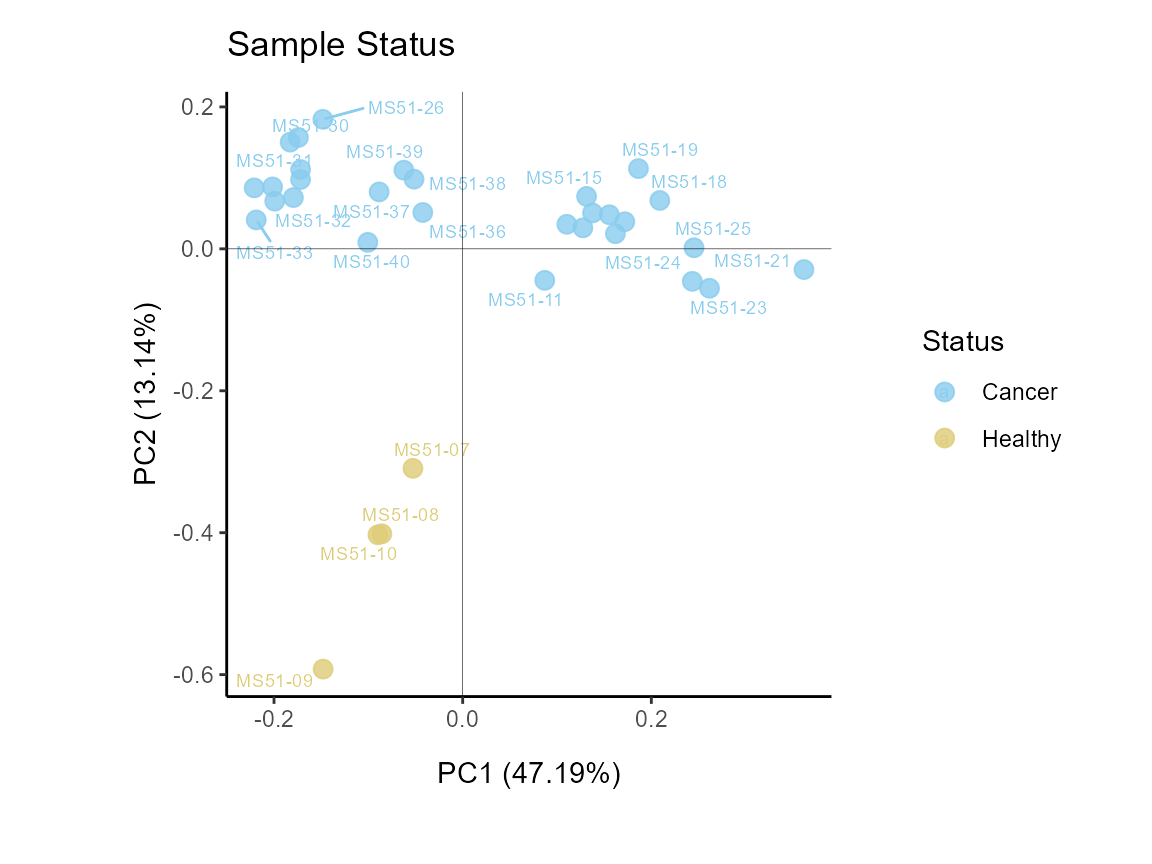
Figure: Do the samples cluster for the conditions?
The different cell lines we have are either control or cancerous, so
we can display this too.
MetaProViz::VizPCA(SettingsInfo= c(color="Status"),
SettingsFile_Sample = MetaData_Sample ,
InputData=Input_PCA,
PlotName = "Sample Status")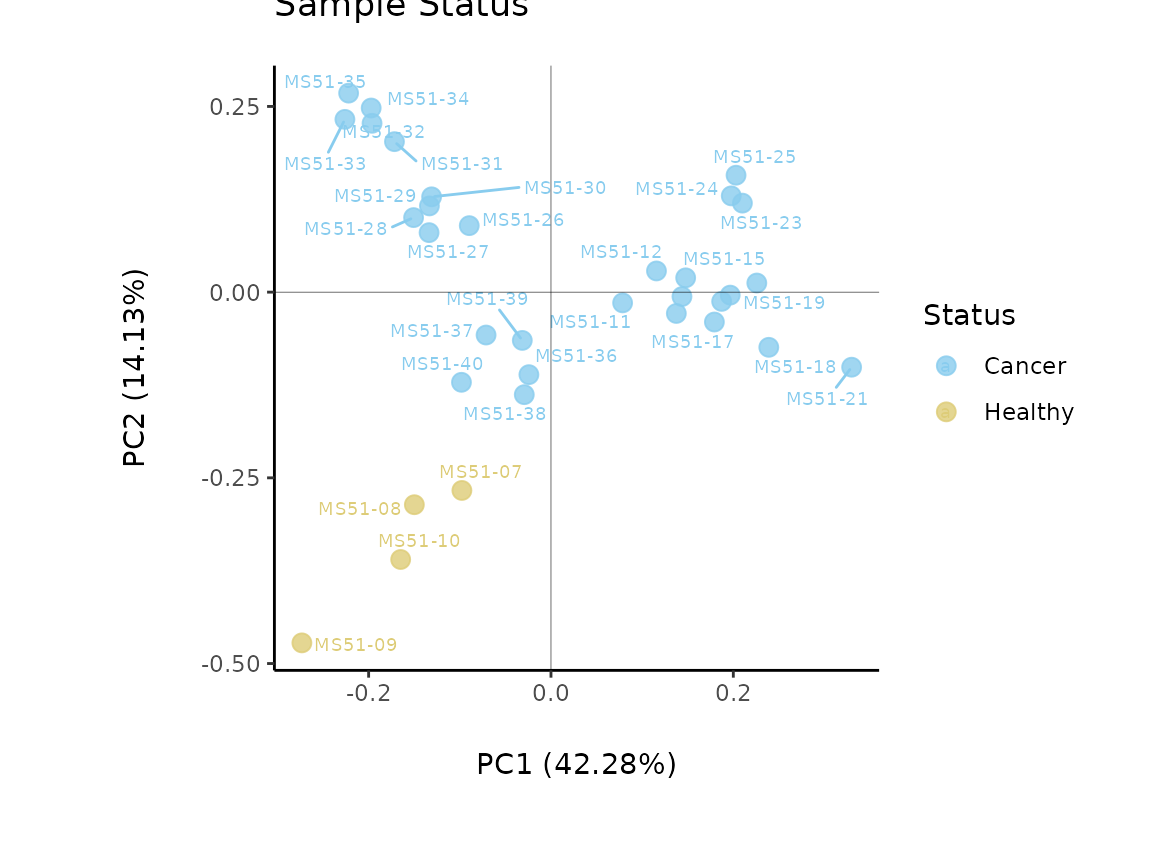
Figure: Do the samples cluster for the Cell status?
Heatmaps
Clustered heatmaps can be useful to understand the patterns in the
data, which will be showcased on different examples.
As input, we need a DF that contains the samples as rownames and the
features (=metabolites) as column names:
Input_Heatmap <- Media_Preprocessed[,-c(1:4)] #remove columns that include Metadata such as cell type,...| valine-d8 | hipppuric acid-d5 | 2-hydroxyglutarate | 2-ketoglutarate | 3-Dehydro-L-threonate | |
|---|---|---|---|---|---|
| MS51-07 | 5049281744 | 30218506574 | -868601575 | 1530912430 | 28673393701 |
| MS51-08 | -1441406385 | -11561761745 | 1108346025 | 3684365990 | 39313101752 |
| MS51-09 | 2563904070 | -10237061776 | -189633550 | -9097094735 | -67782920598 |
| MS51-10 | -6171779428 | -8419683053 | -50110899 | 3881816314 | -203574855 |
| MS51-11 | 27378617230 | 123649127347 | 4311754839 | 23286535748 | -16721754062 |
| MS51-12 | 31998154622 | 99965859879 | 808357705 | 19381509727 | -16349278172 |
Now we can generate an overview heatmap. Since we plot all metabolites
the metabolite names are not plotted since this would get too crowded
(You can enforce this by changing the parameter
enforce_FeatureNames = TRUE).
MetaProViz::VizHeatmap(InputData = Input_Heatmap,
PlotName = "Overview")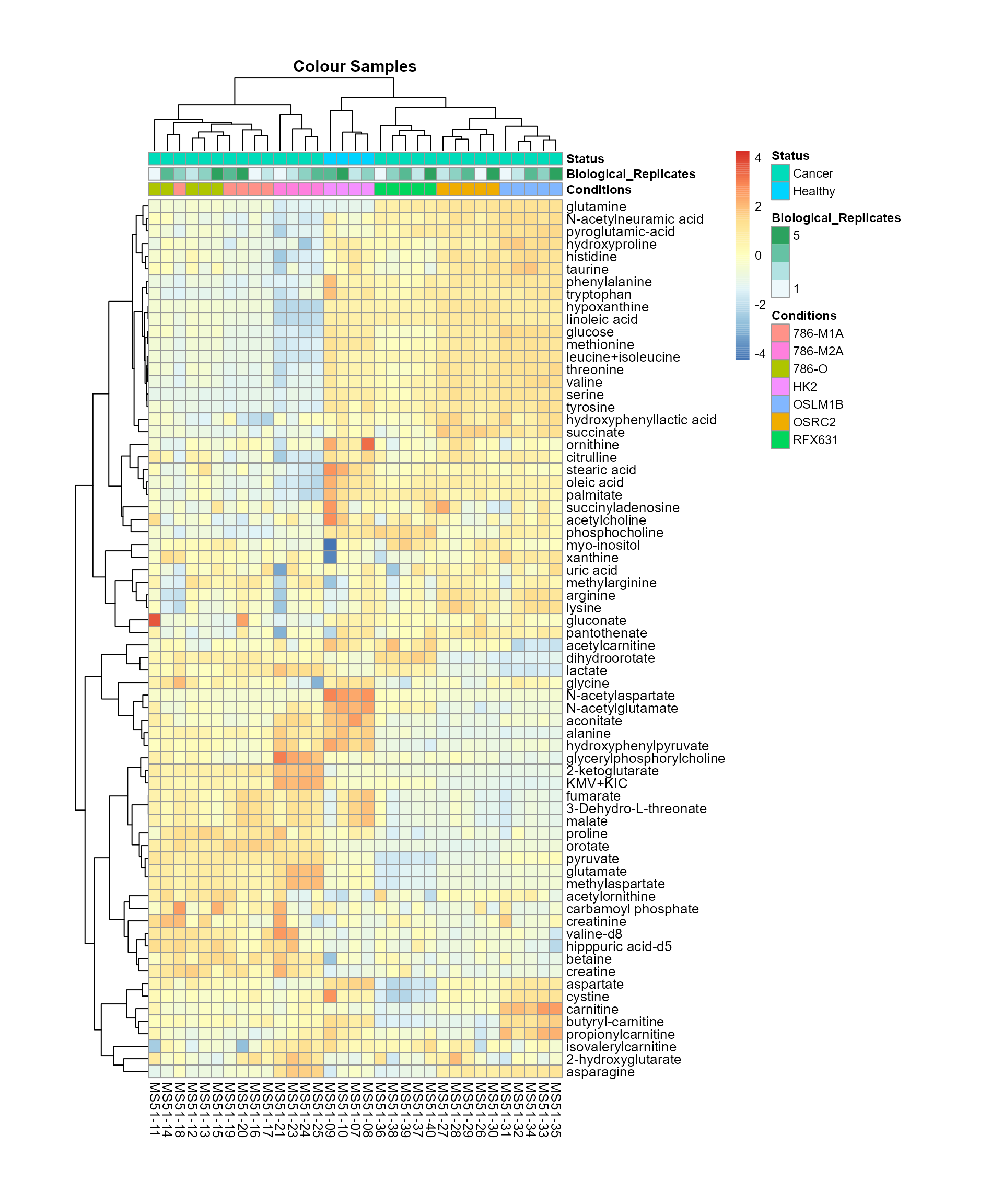
Overview heatmap.
Here we can add as many sample metadata information as needed at the
same time:
MetaProViz::VizHeatmap(InputData = Input_Heatmap,
SettingsFile_Sample = MetaData_Sample,
SettingsInfo = c(color_Sample = list("Conditions","Biological_Replicates", "Status")),
PlotName = "Colour Samples")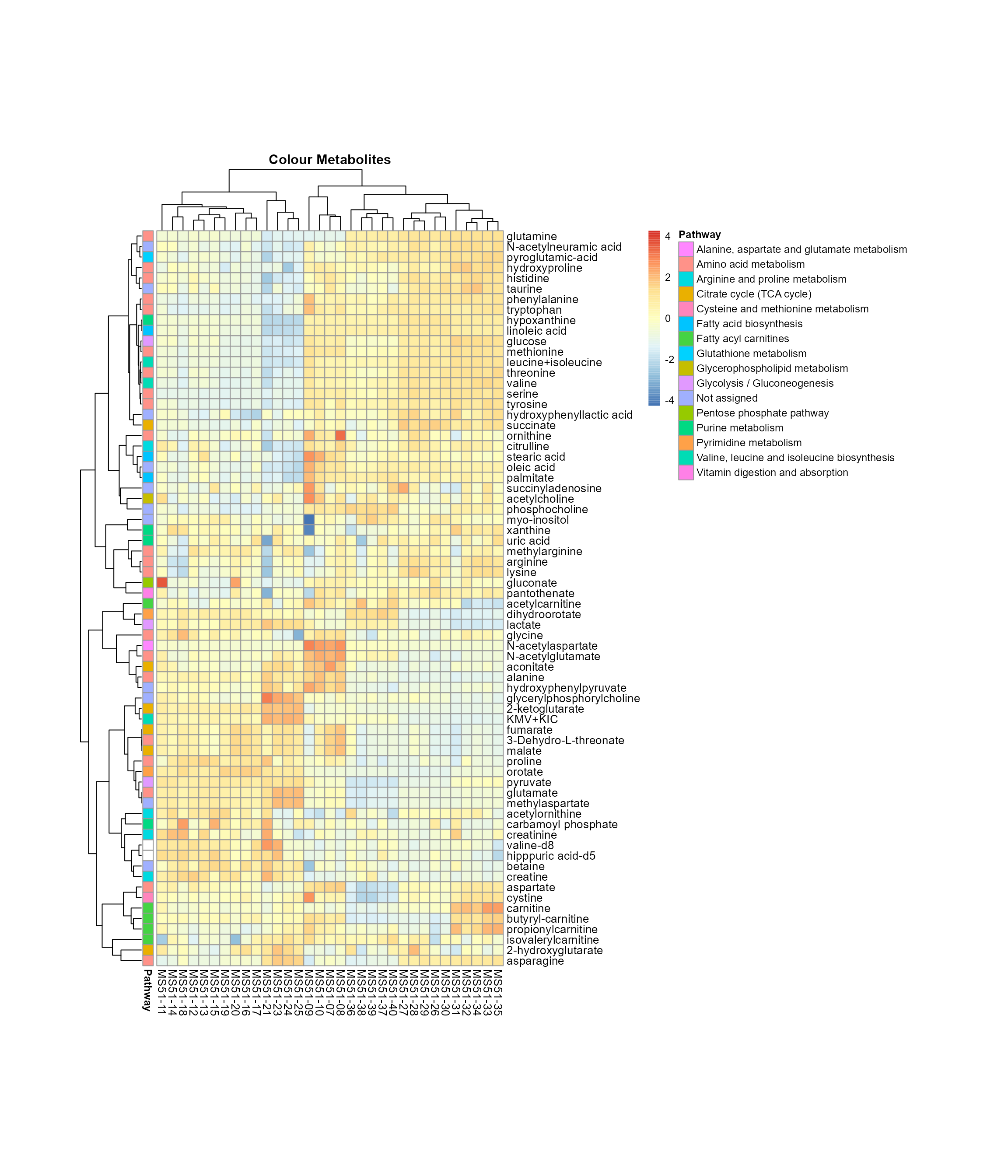
Colour for sample metadata.
Moreover, we can also add metabolite metadata information:
MetaProViz::VizHeatmap(InputData = Input_Heatmap,
SettingsFile_Sample = MetaData_Sample,
SettingsInfo = c(color_Metab = list("Pathway")),
SettingsFile_Metab = MappingInfo,
PlotName = "Colour Metabolites")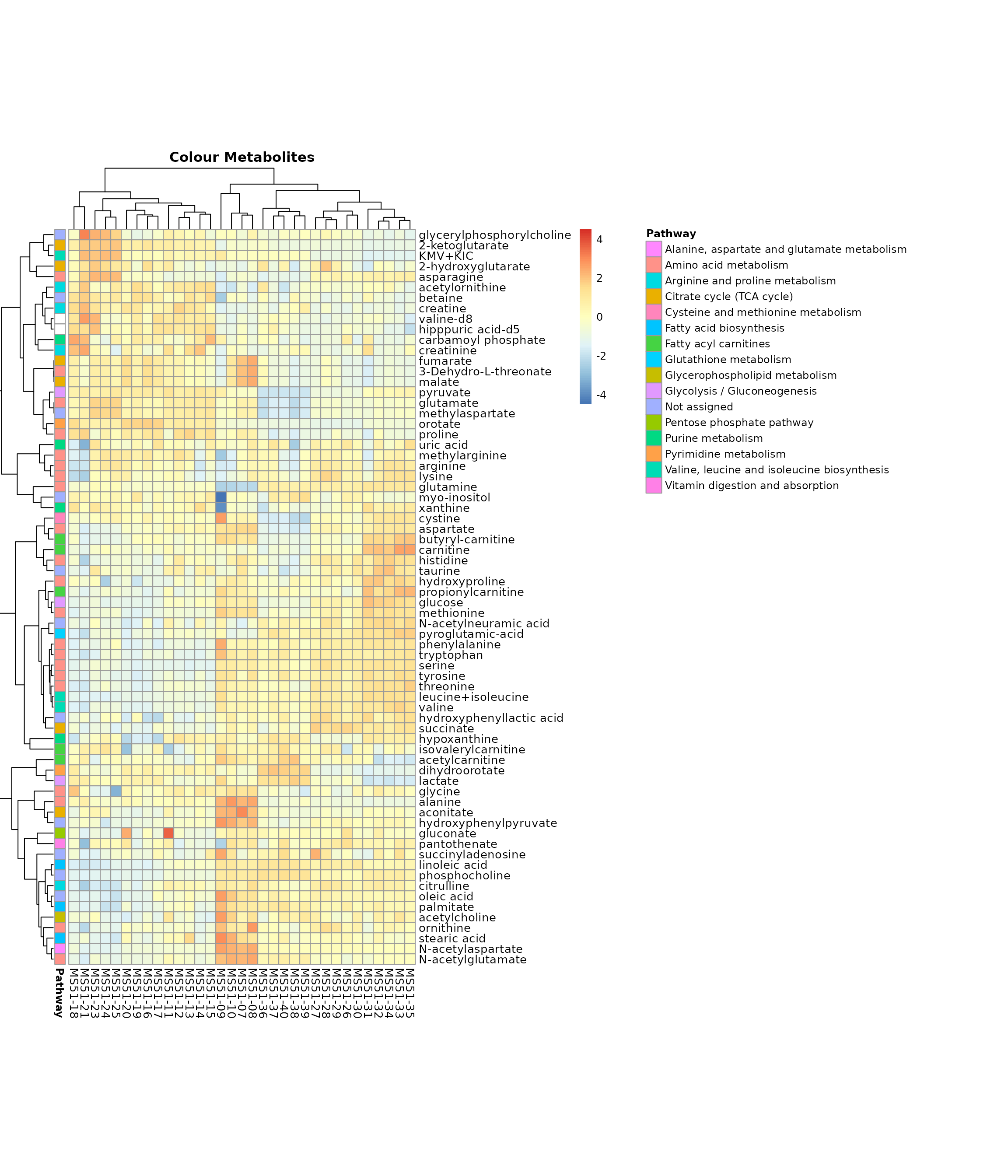
Colour for metabolite metadata.
Lastly, by generate individual plot for e.g. each pathway or the
metabolite clusters by adding individual (individual_Sample
or individual_Metab) to Plot_SettingsInfo. At
the same time we can still maintain the metadata information for both,
the samples and the metabolites. Together this can help us to draw
biological conclusions about the different pathways: Indeed, we can
observe for the D-Amino acid metabolism many metabolites
fall into the MCA-Cluster Core_DOWN, meaning in comparison
to HK2 cells we have a negative Log2FC for 786-O and 786-M1A.
# individual: One individual plot for each pathway, col annotation: Colour for samples
MetaProViz::VizHeatmap(InputData = Input_Heatmap,
SettingsFile_Sample = MetaData_Sample,
SettingsInfo = c(individual_Metab = "Pathway",
color_Sample = list("Conditions","Biological_Replicates"),
color_Metab = list("Pathway")),
SettingsFile_Metab = MetaData_Metab,
PlotName = "Pathway")
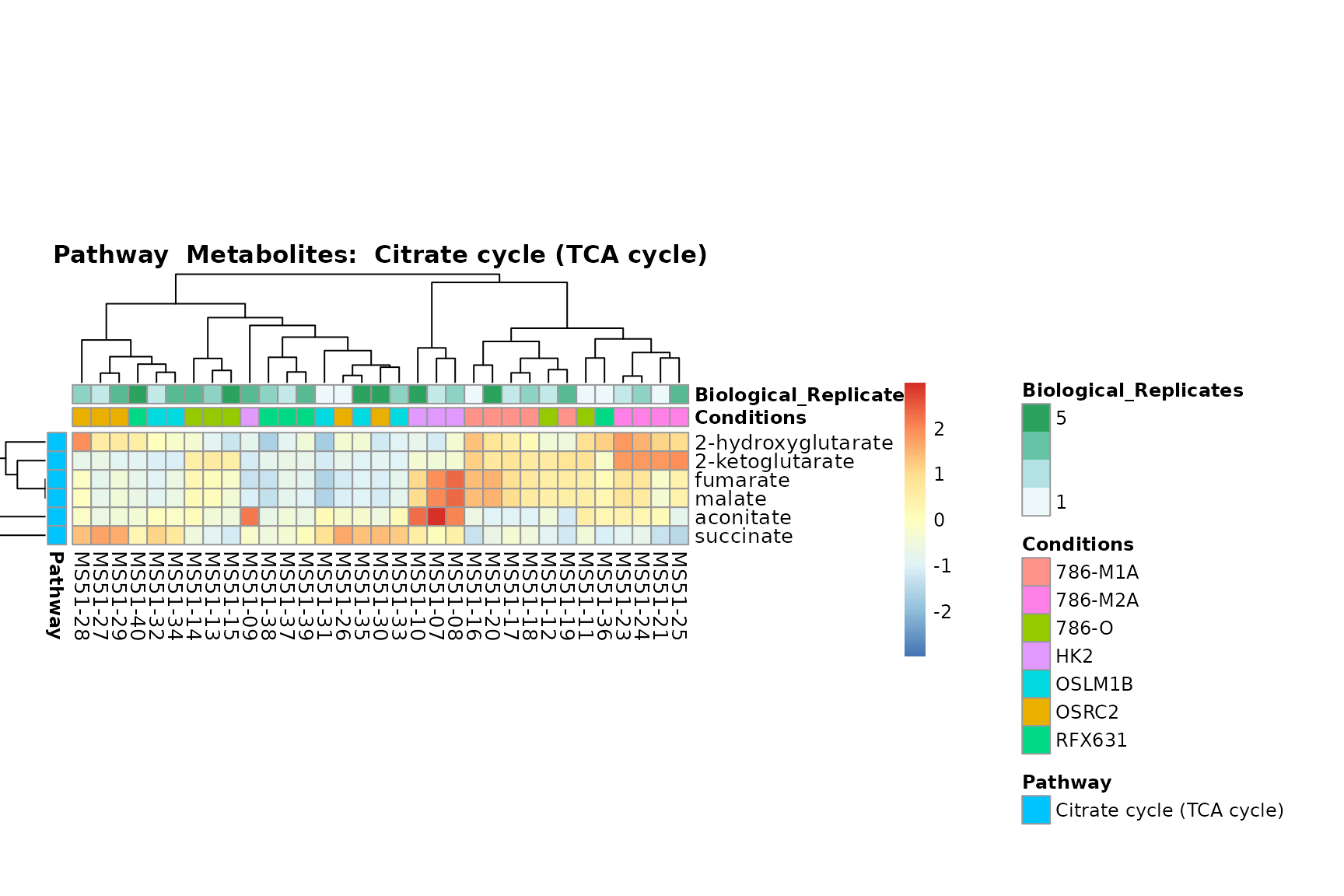
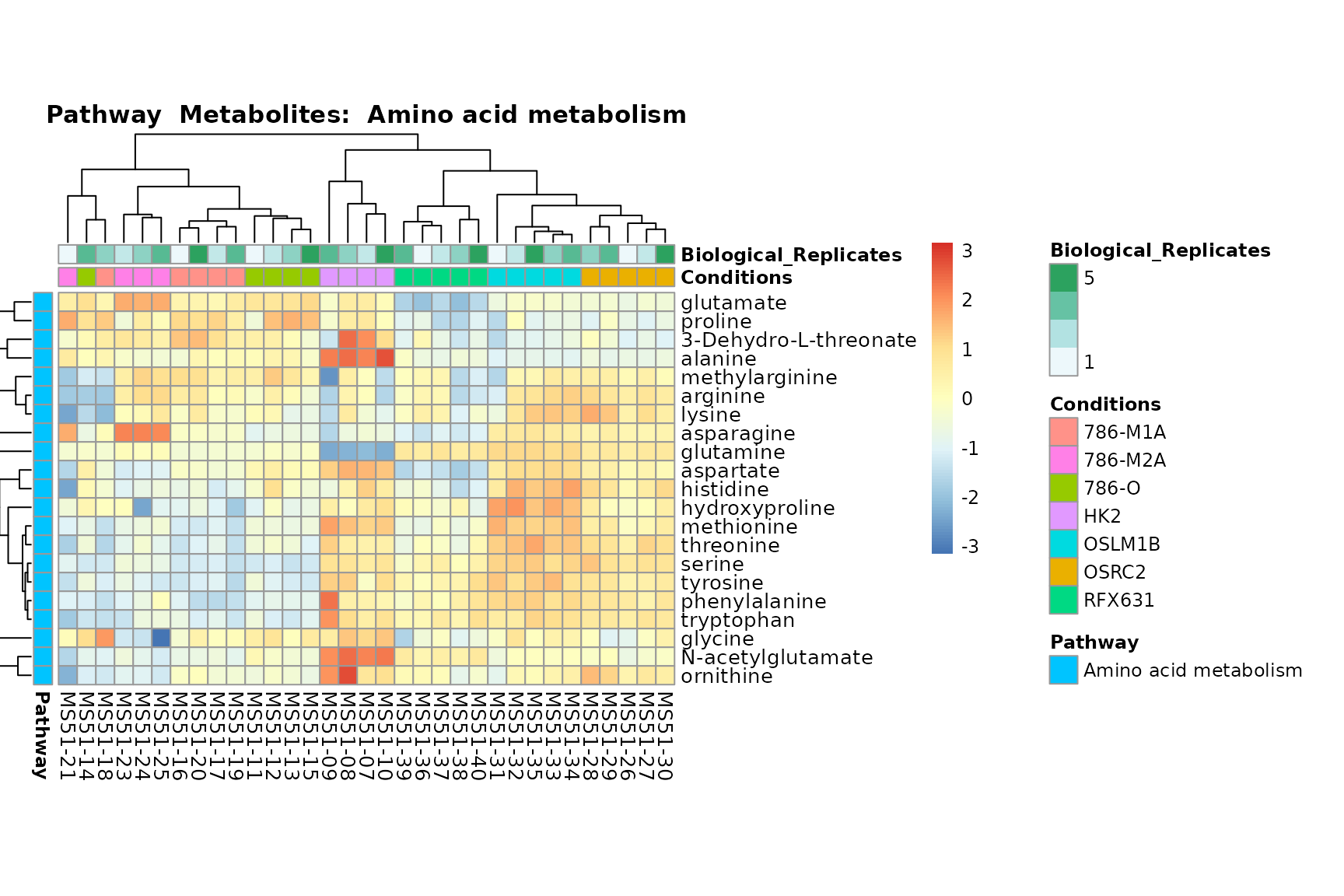
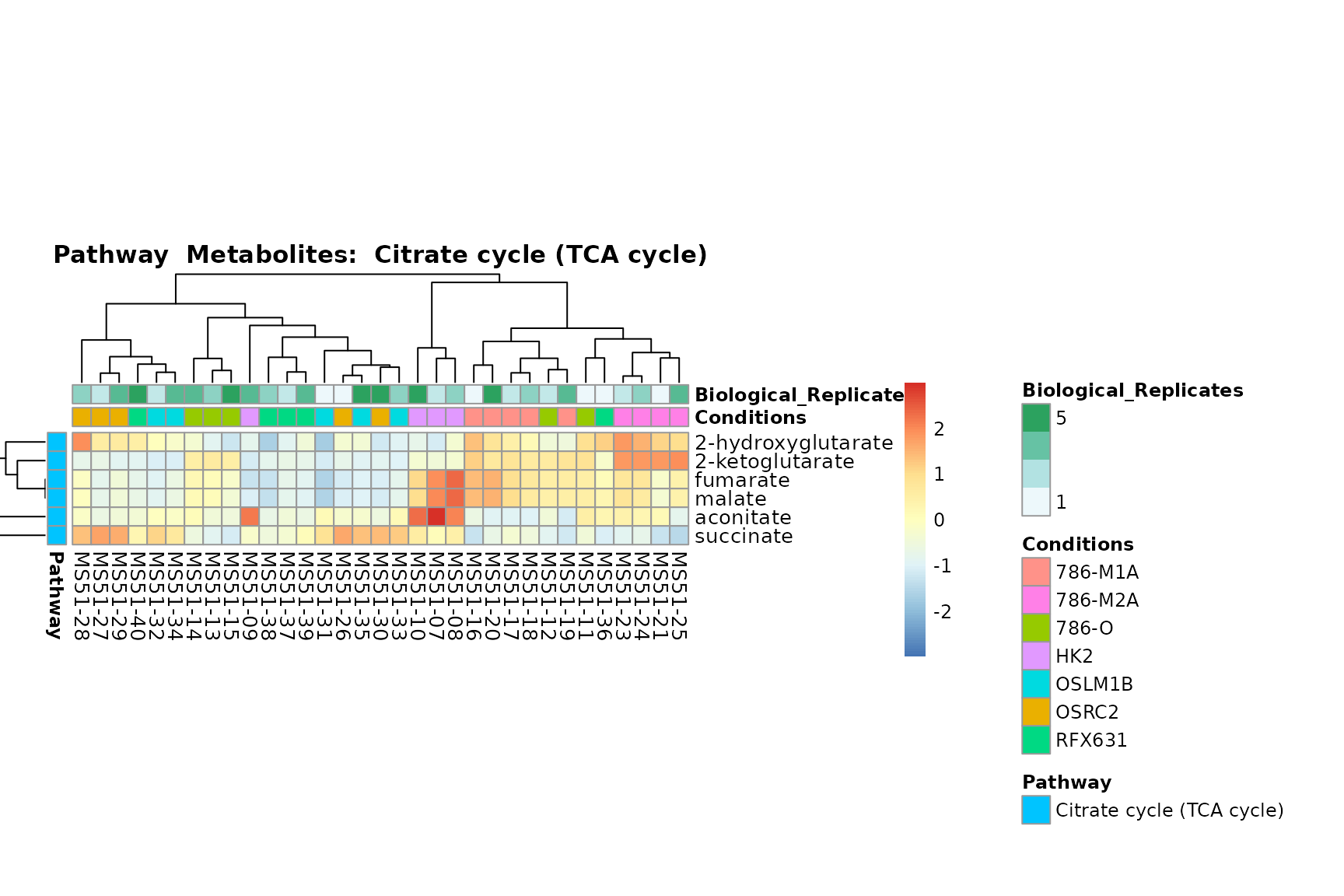

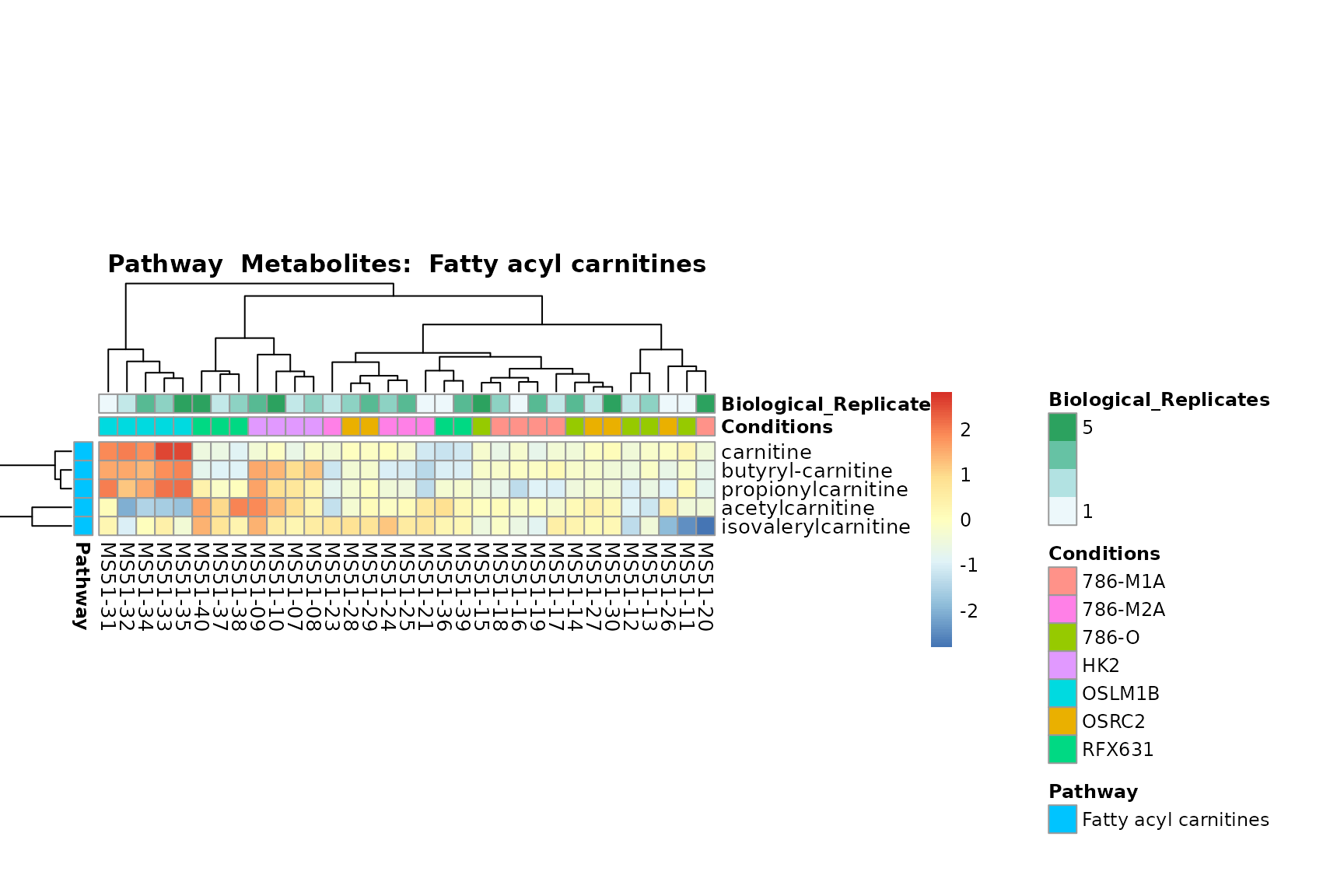
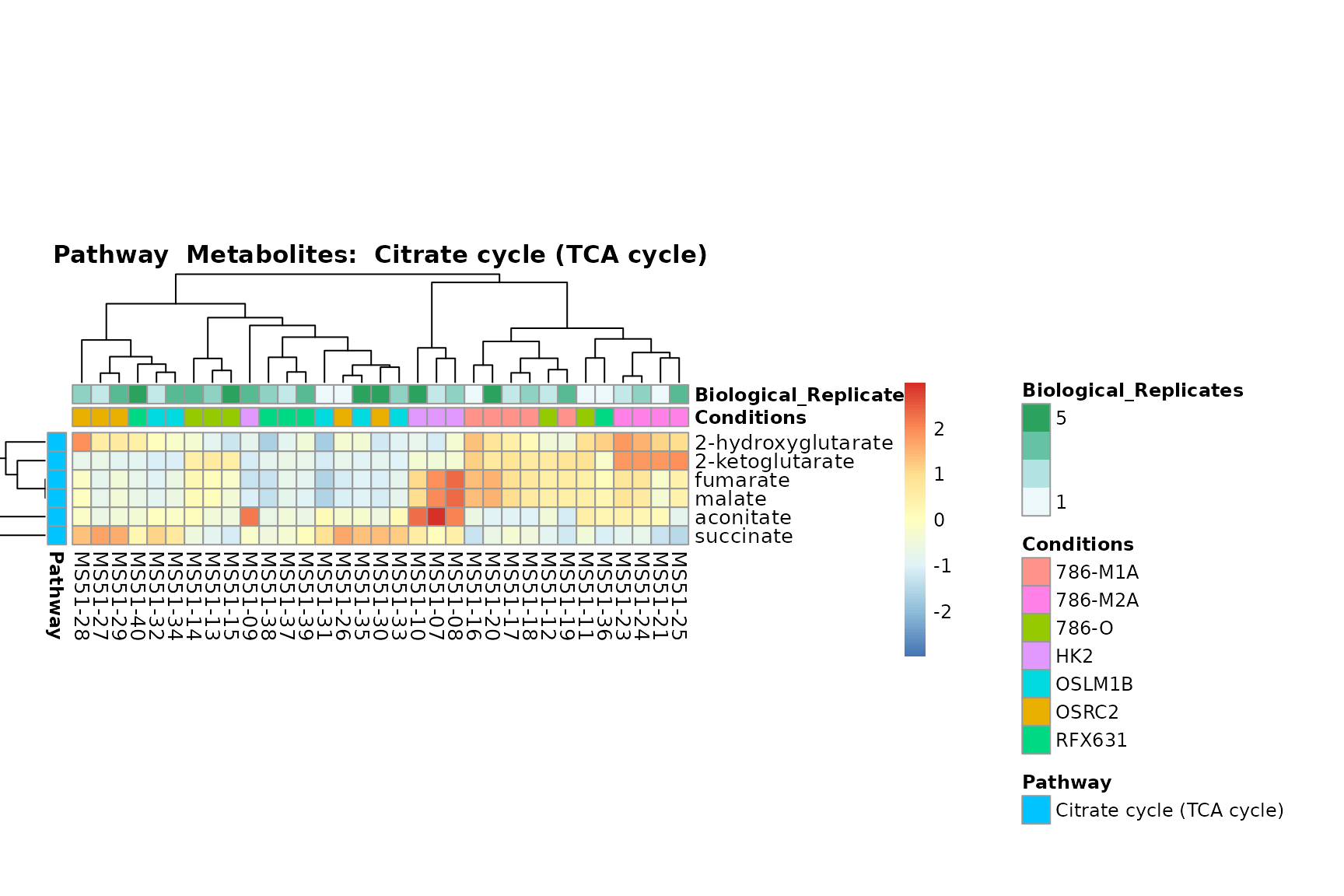
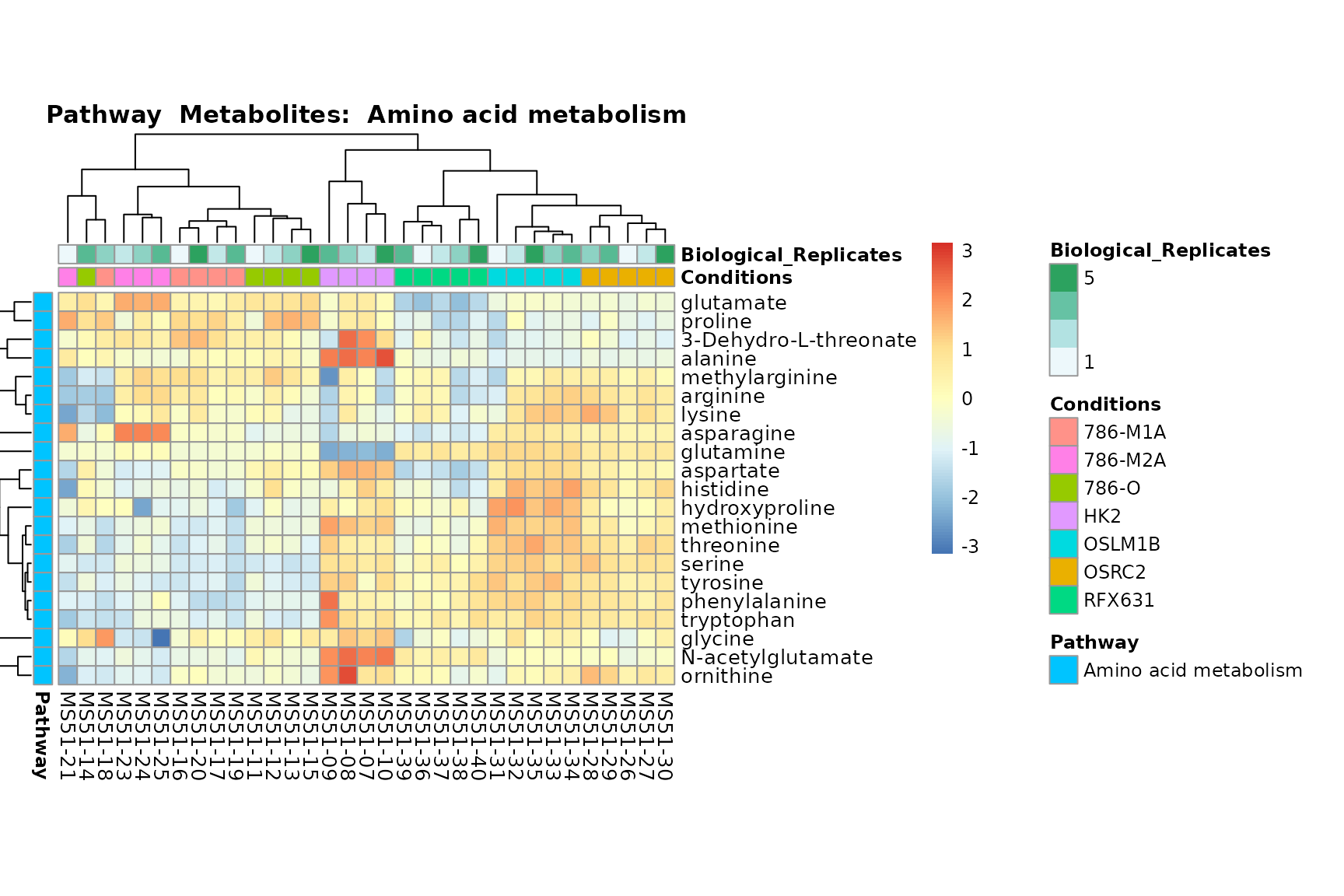

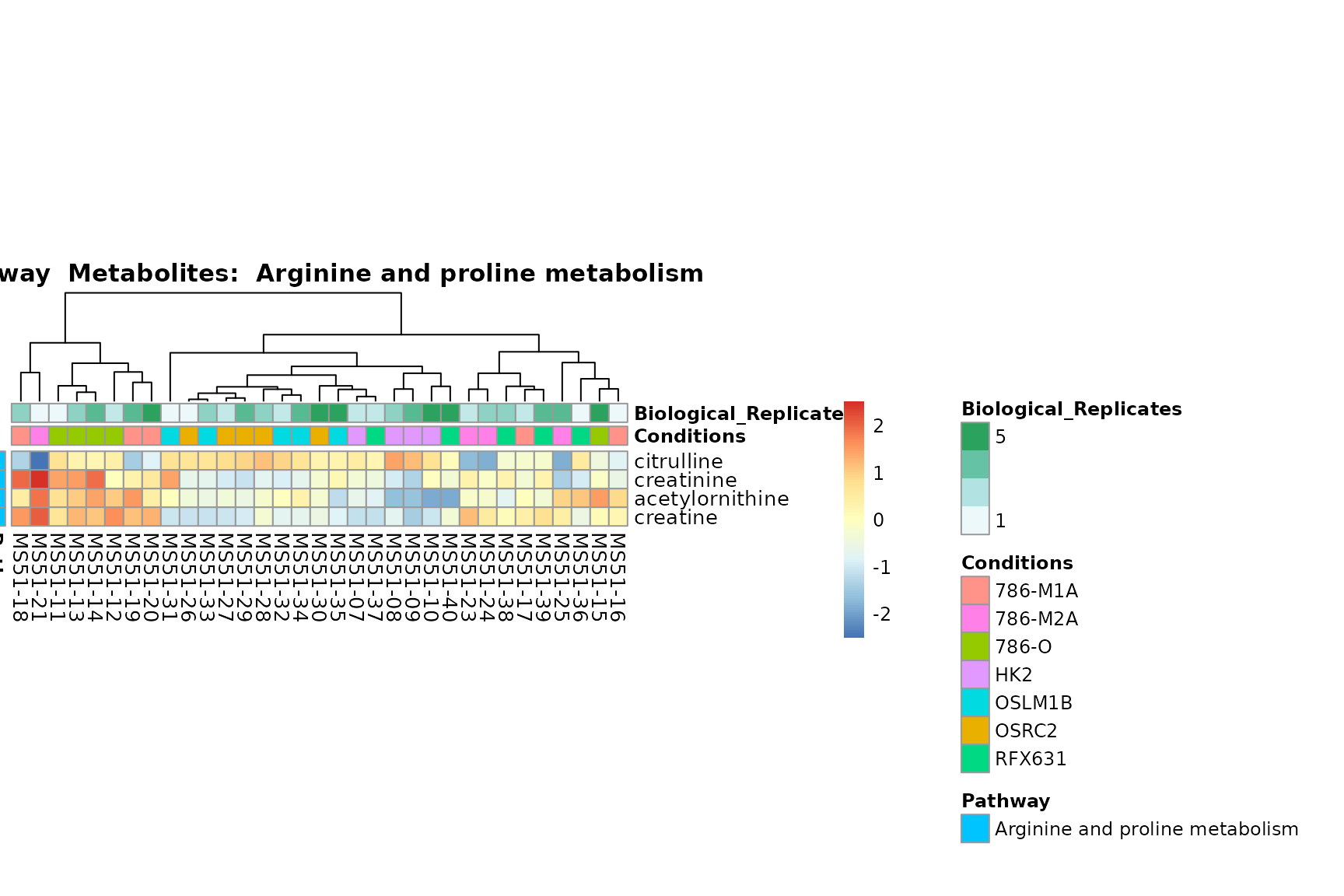
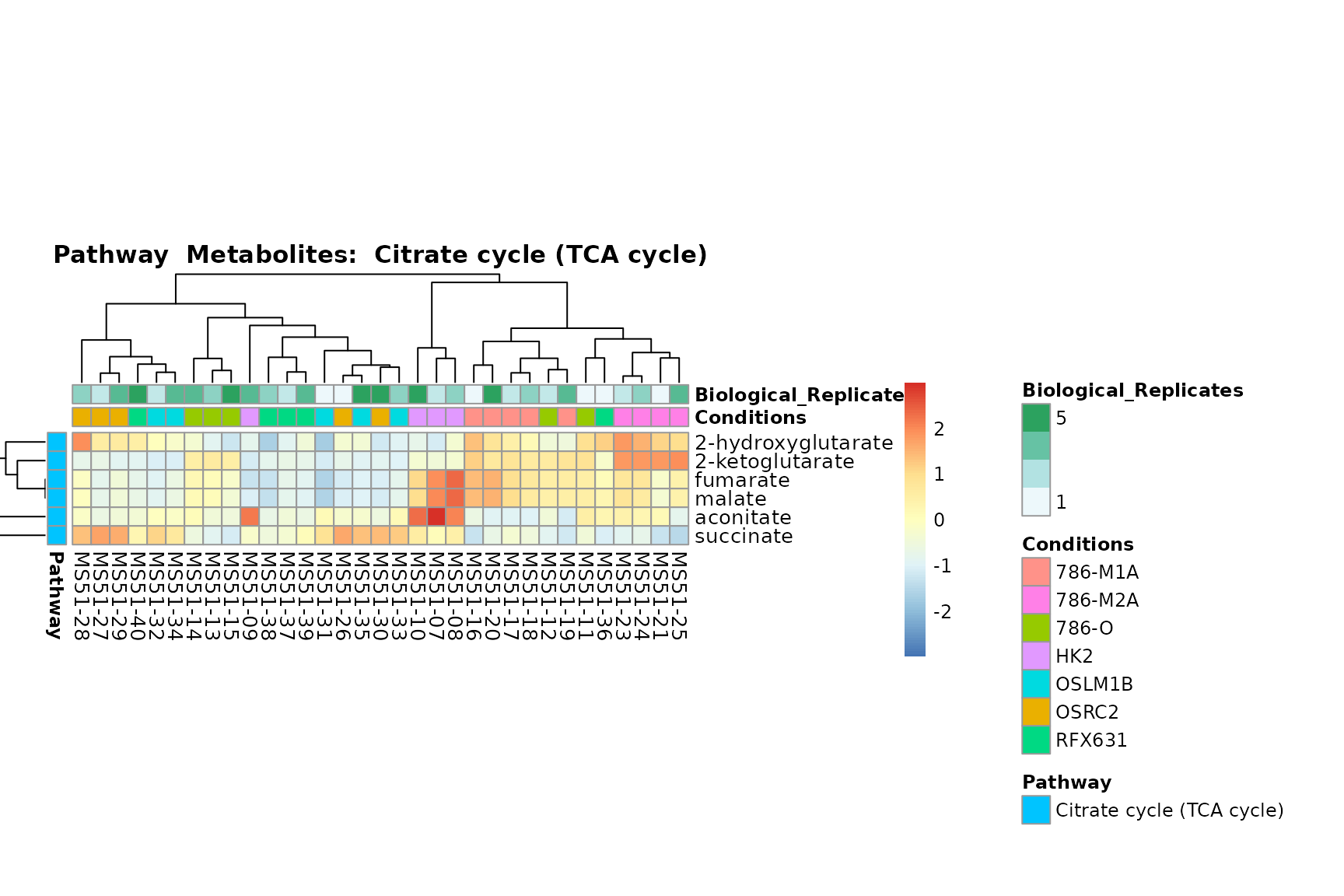
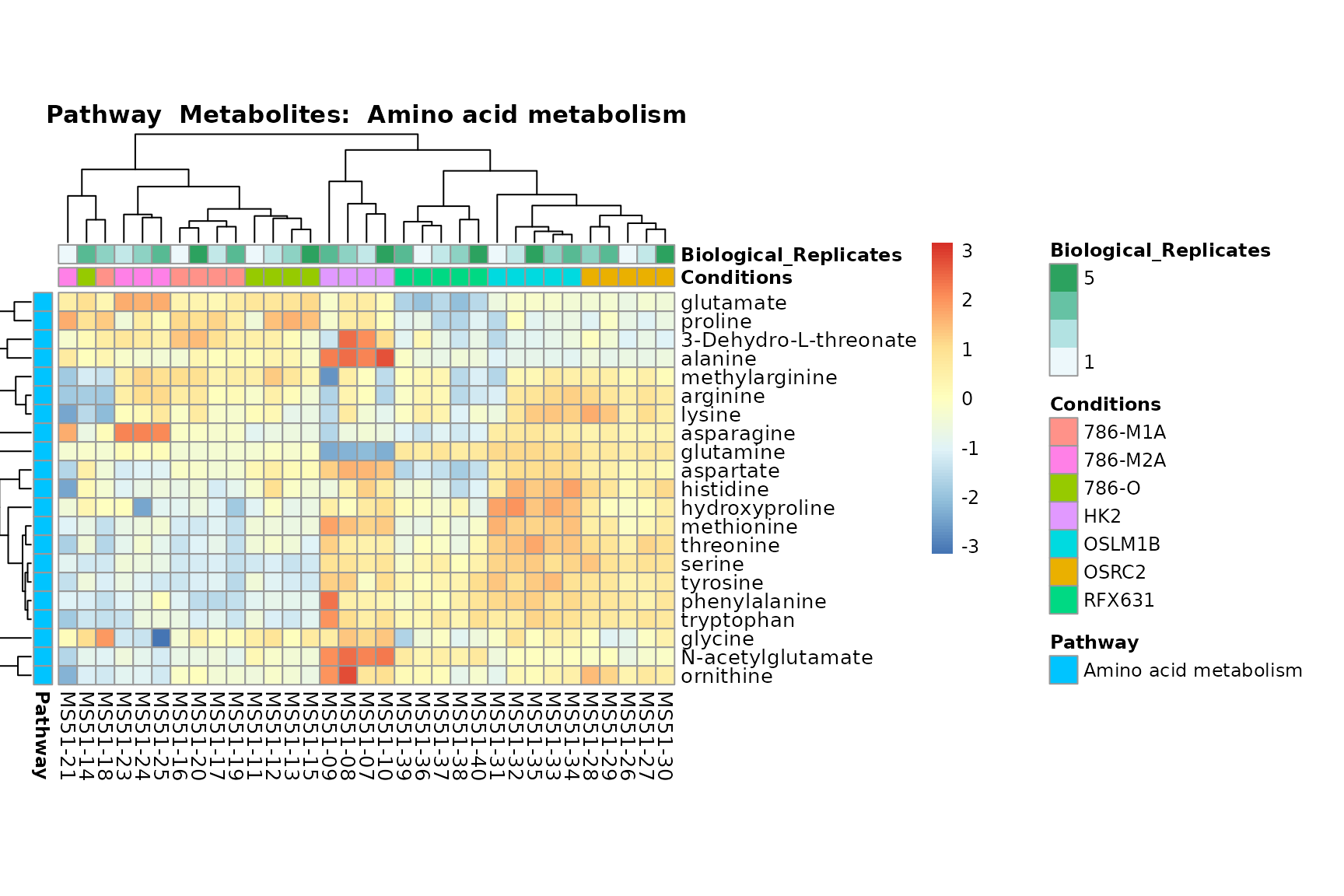
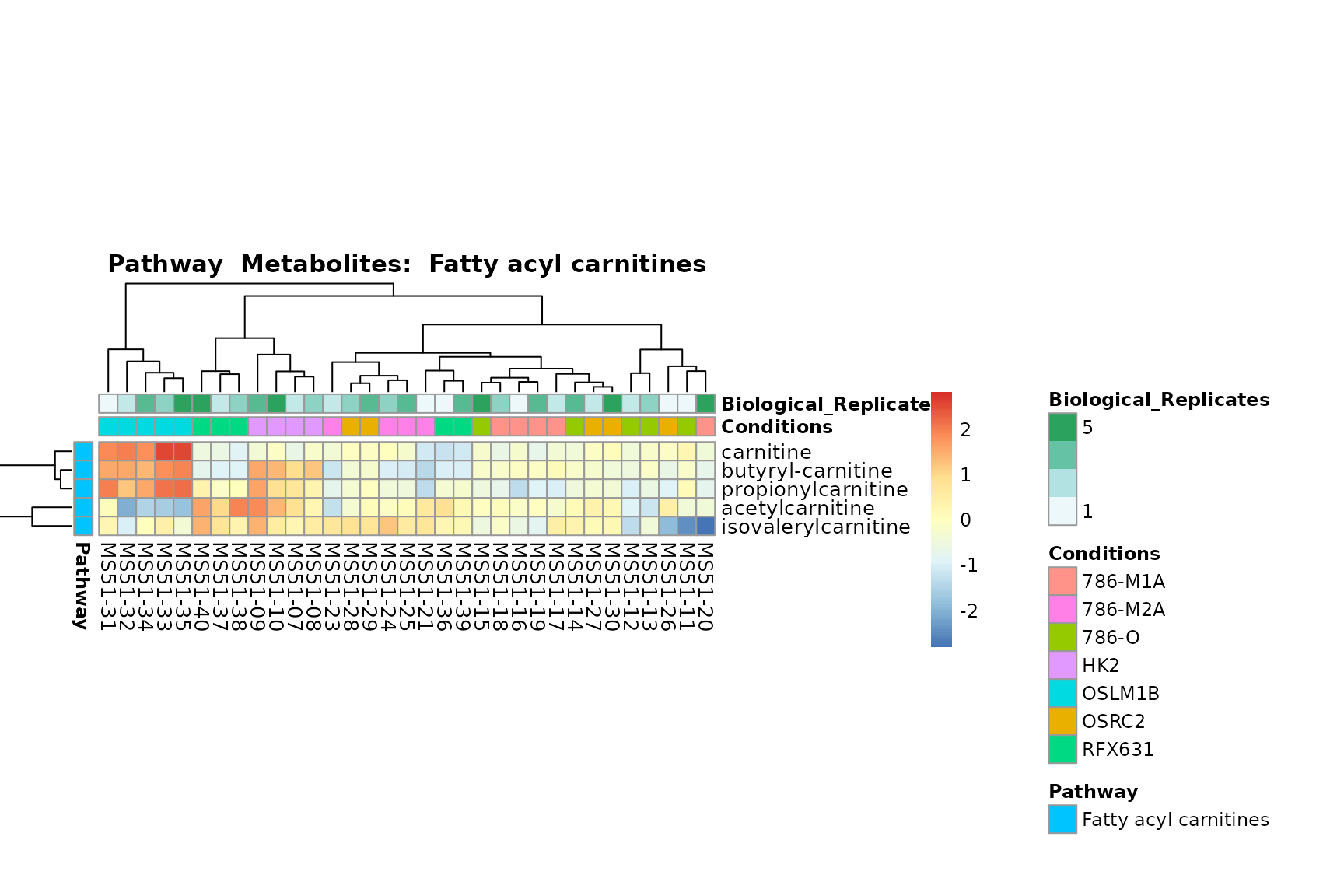

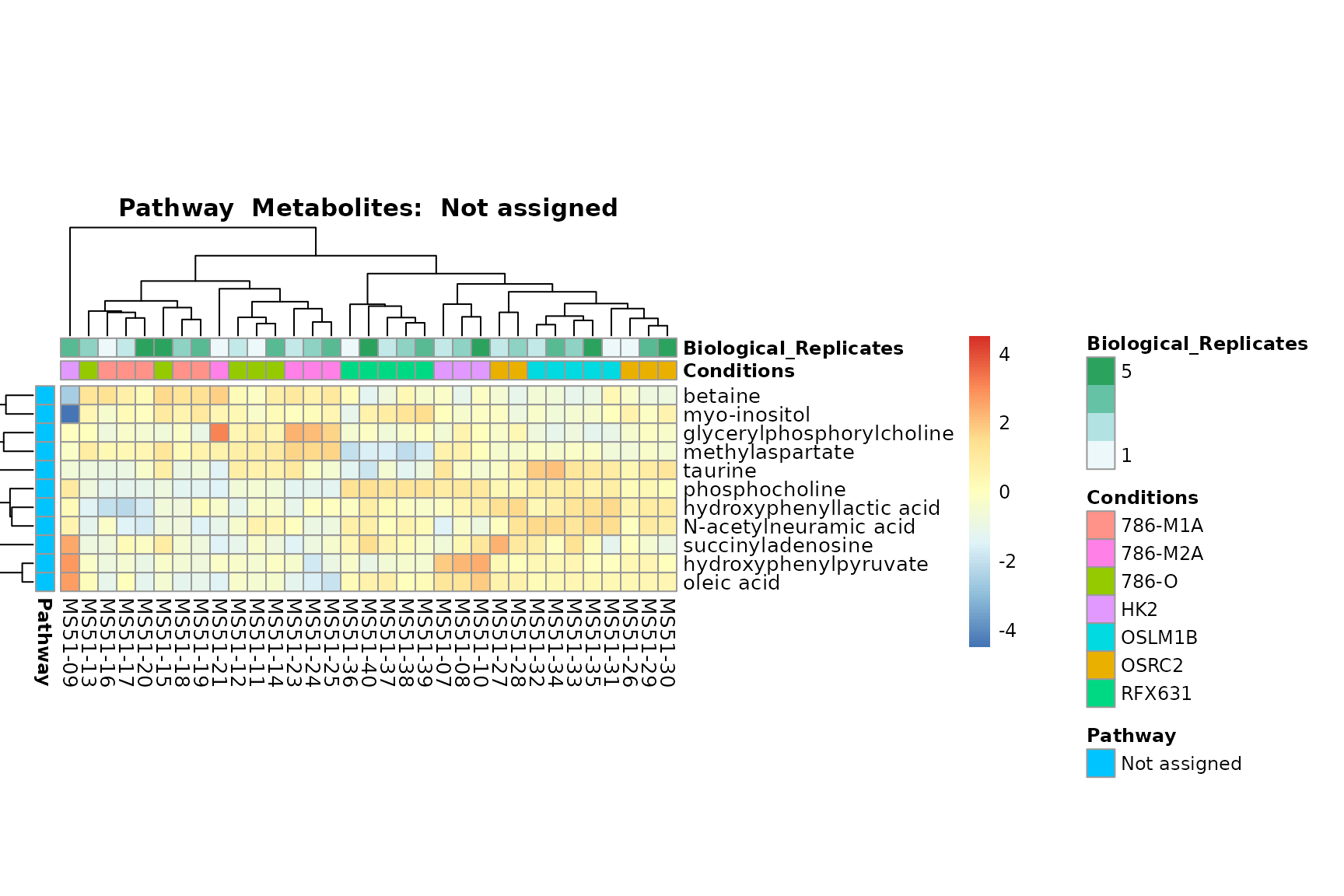
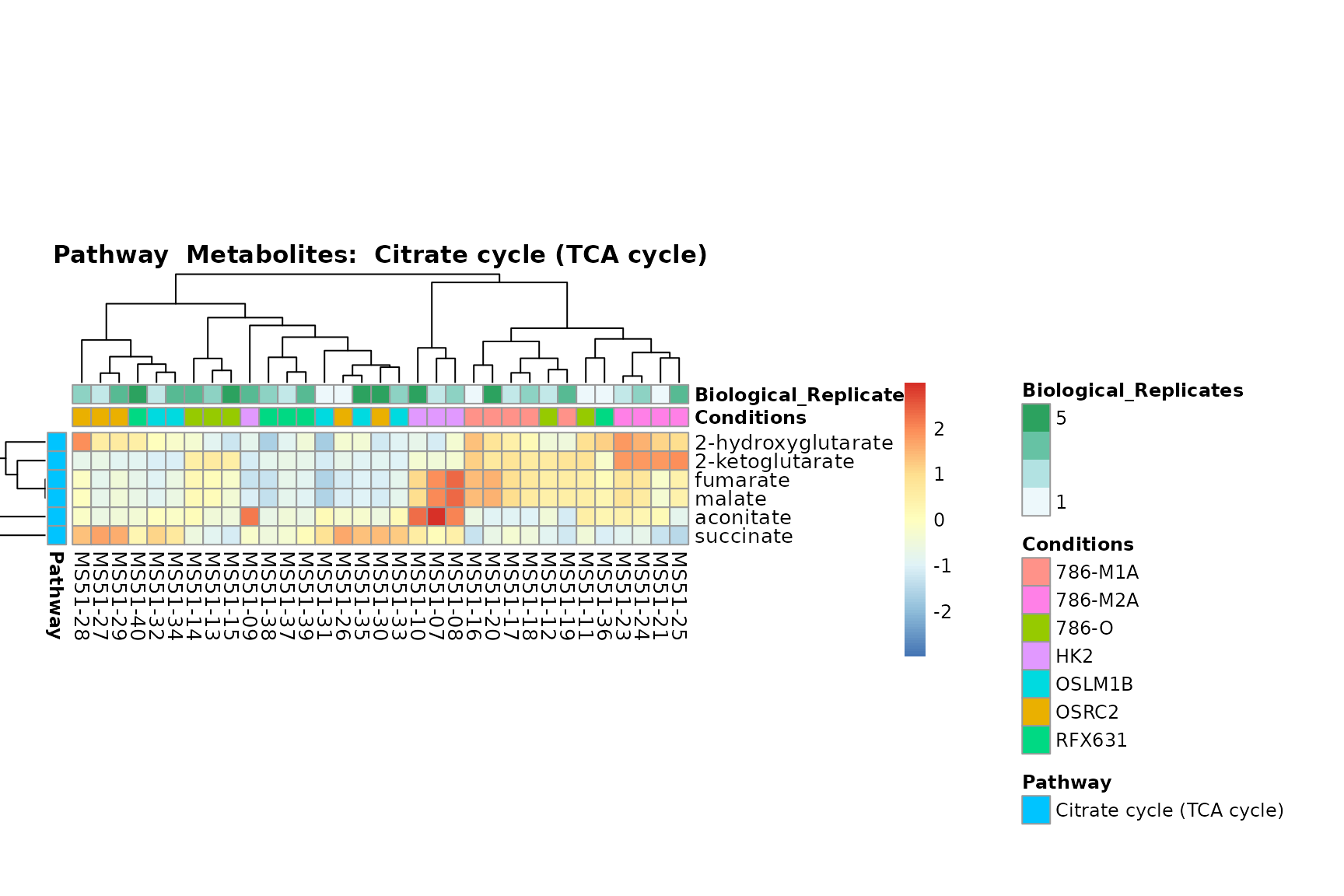

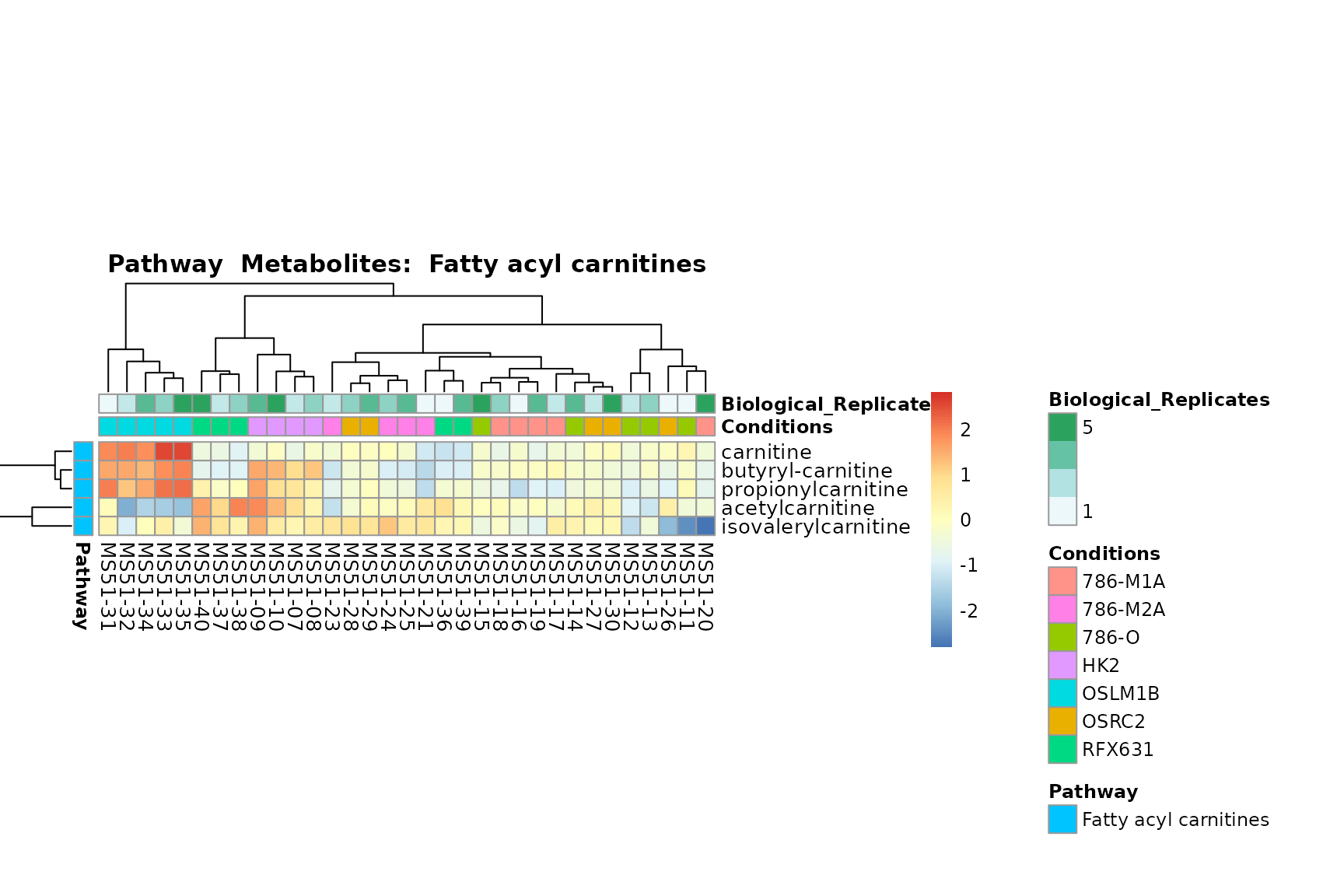
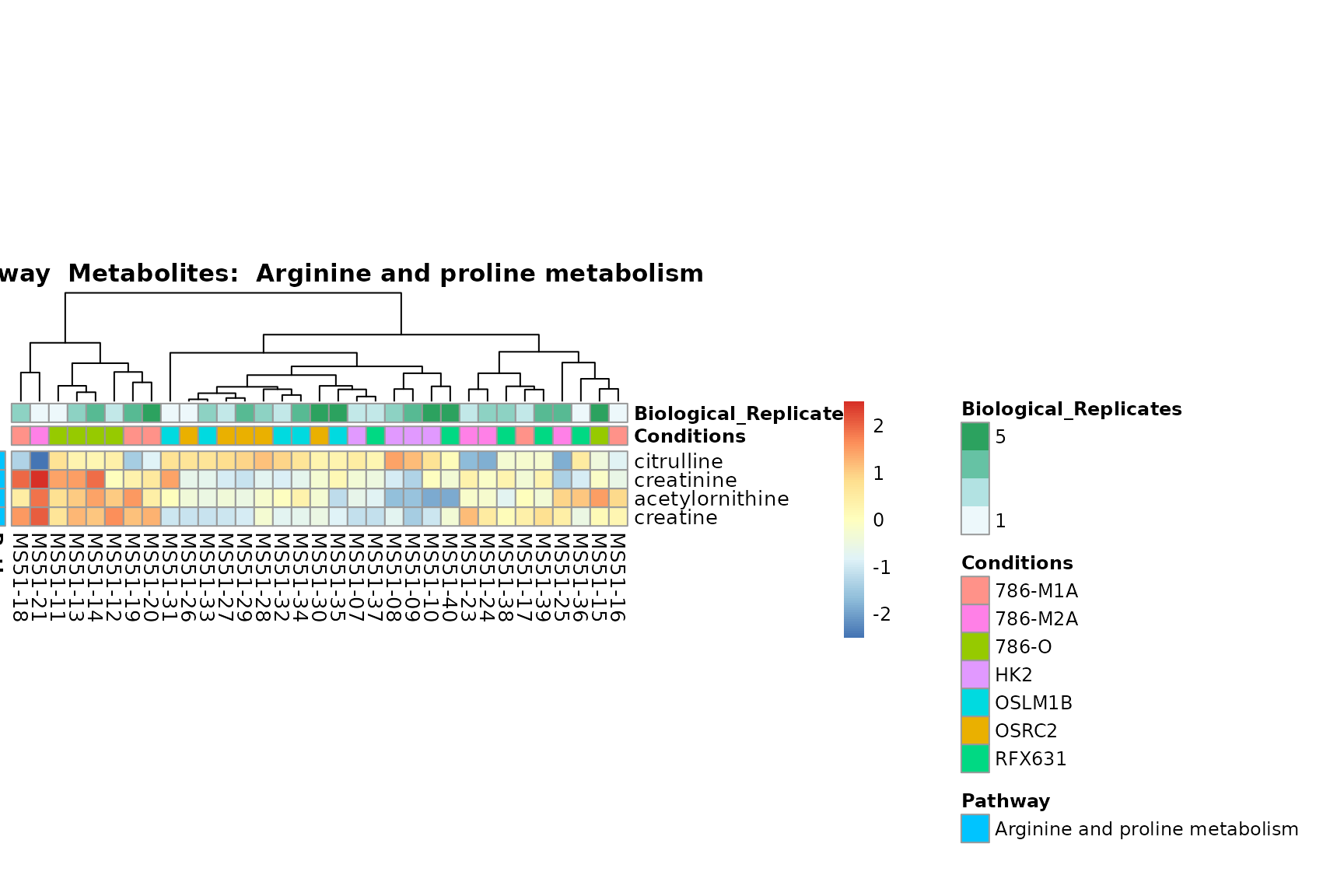
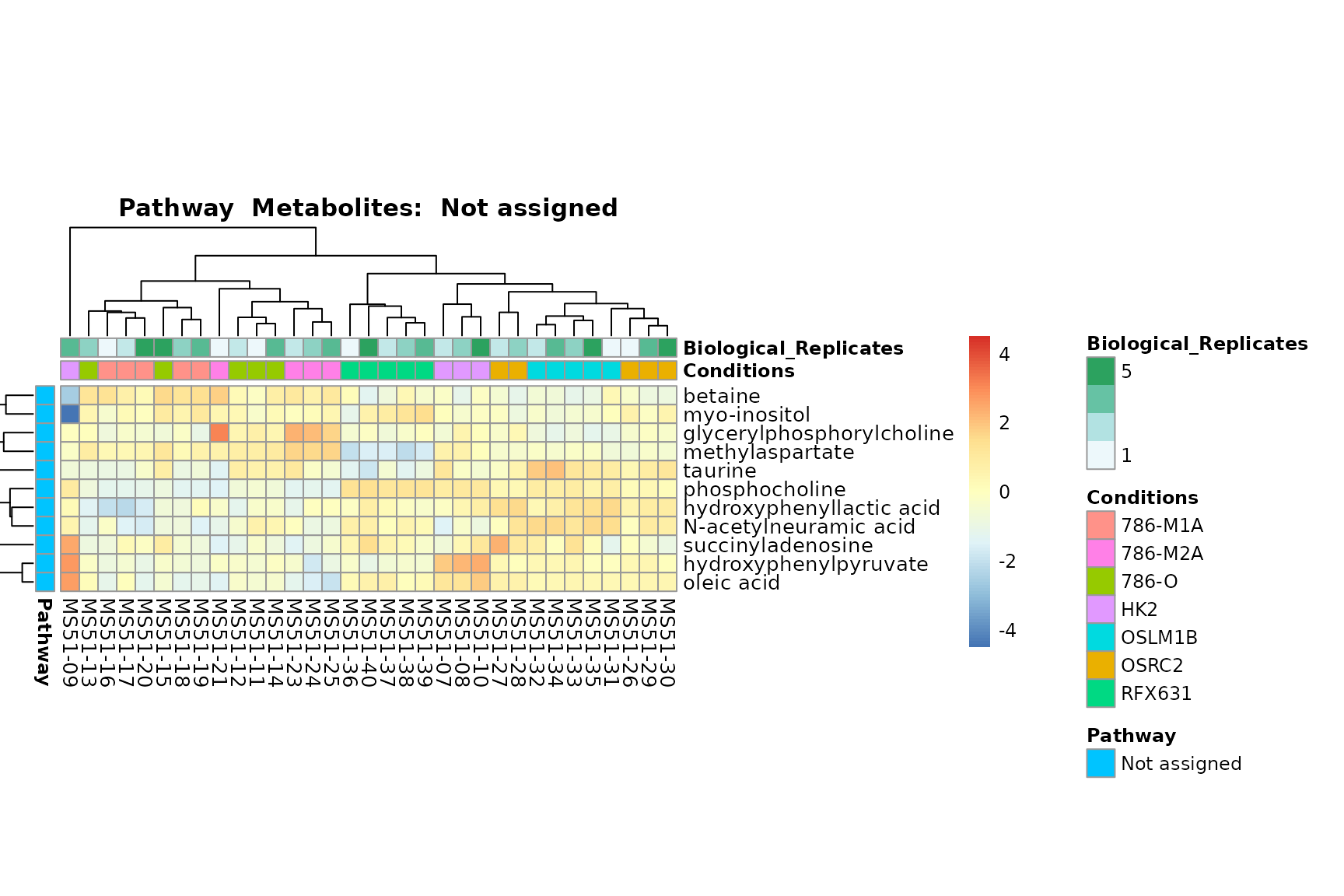
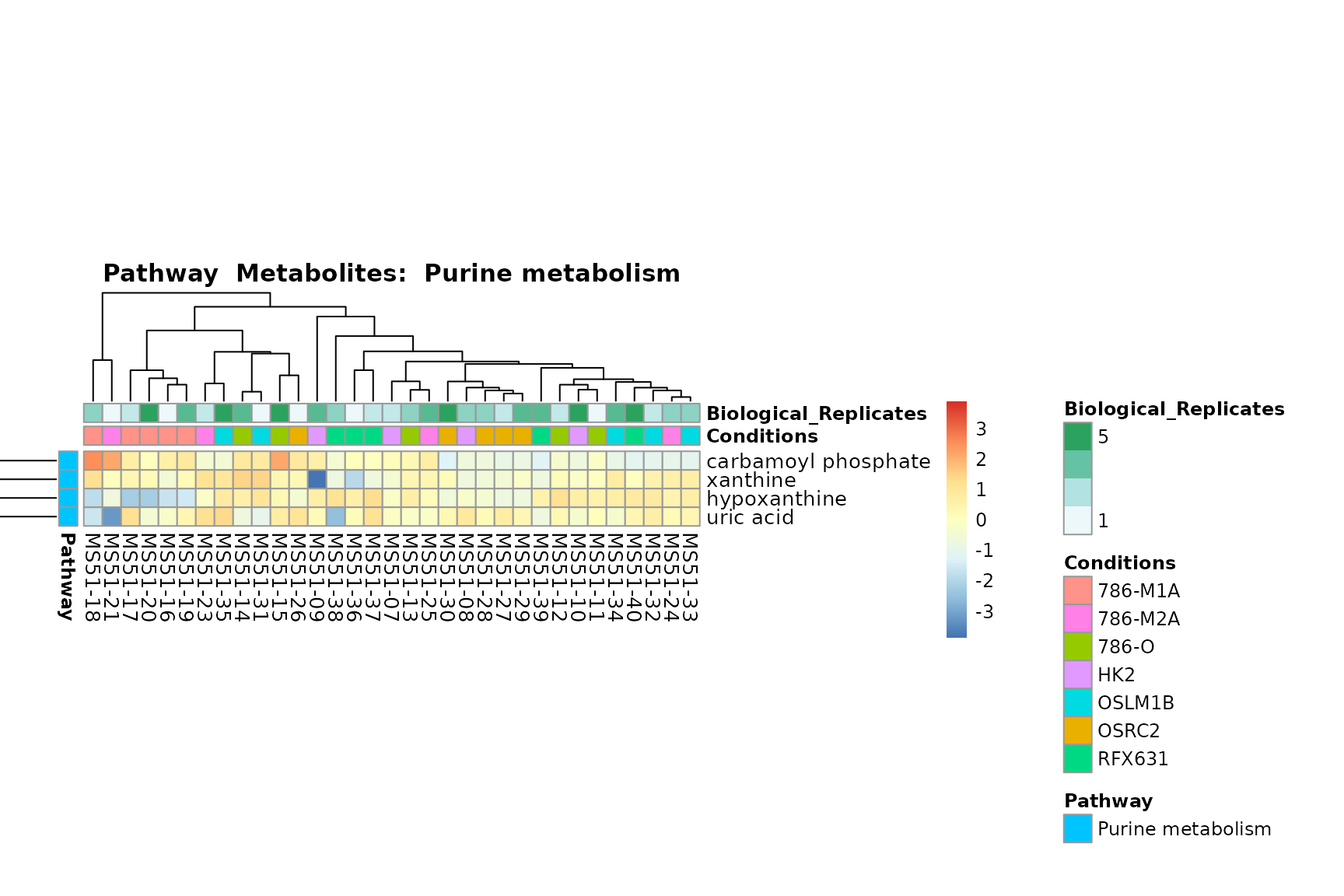
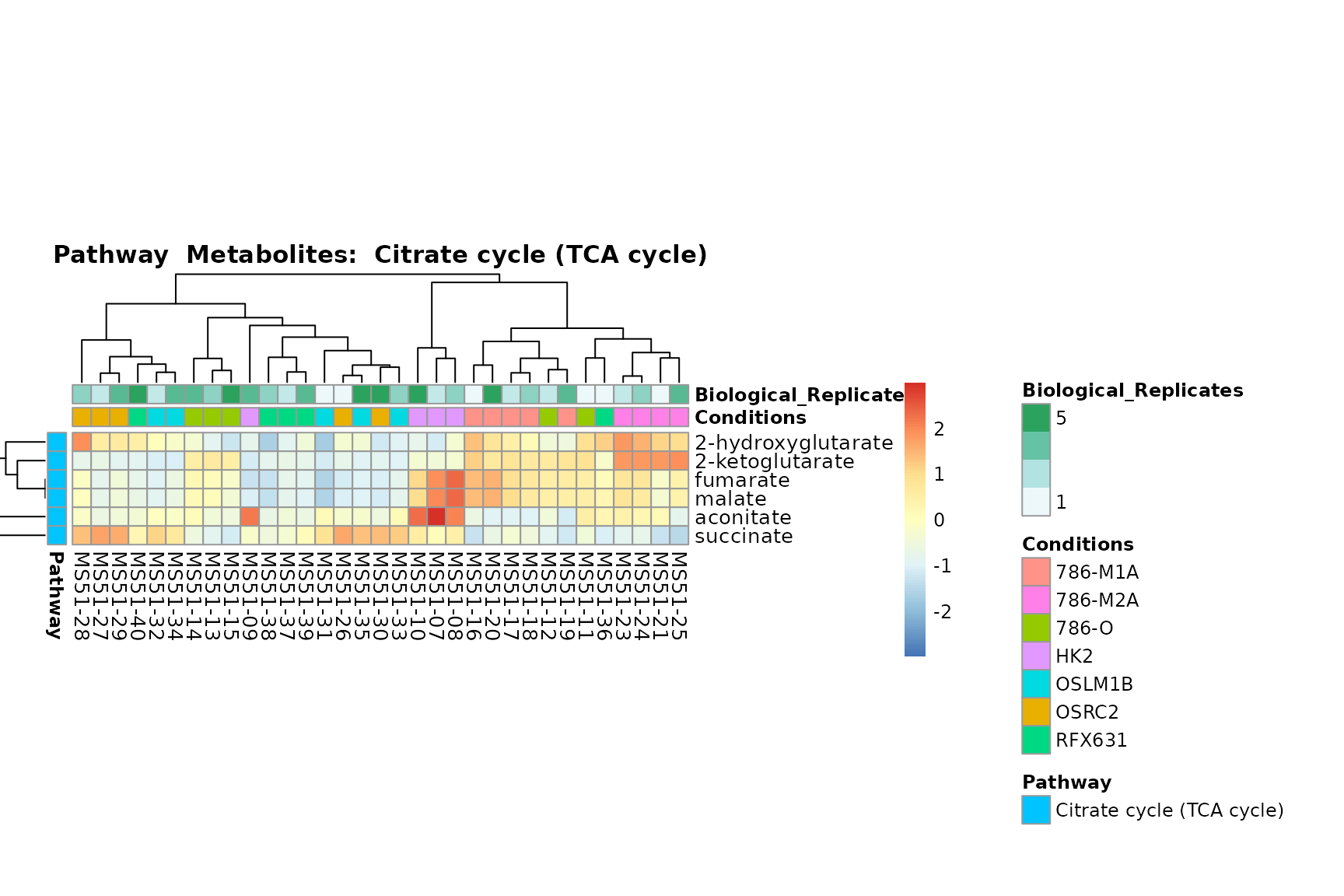

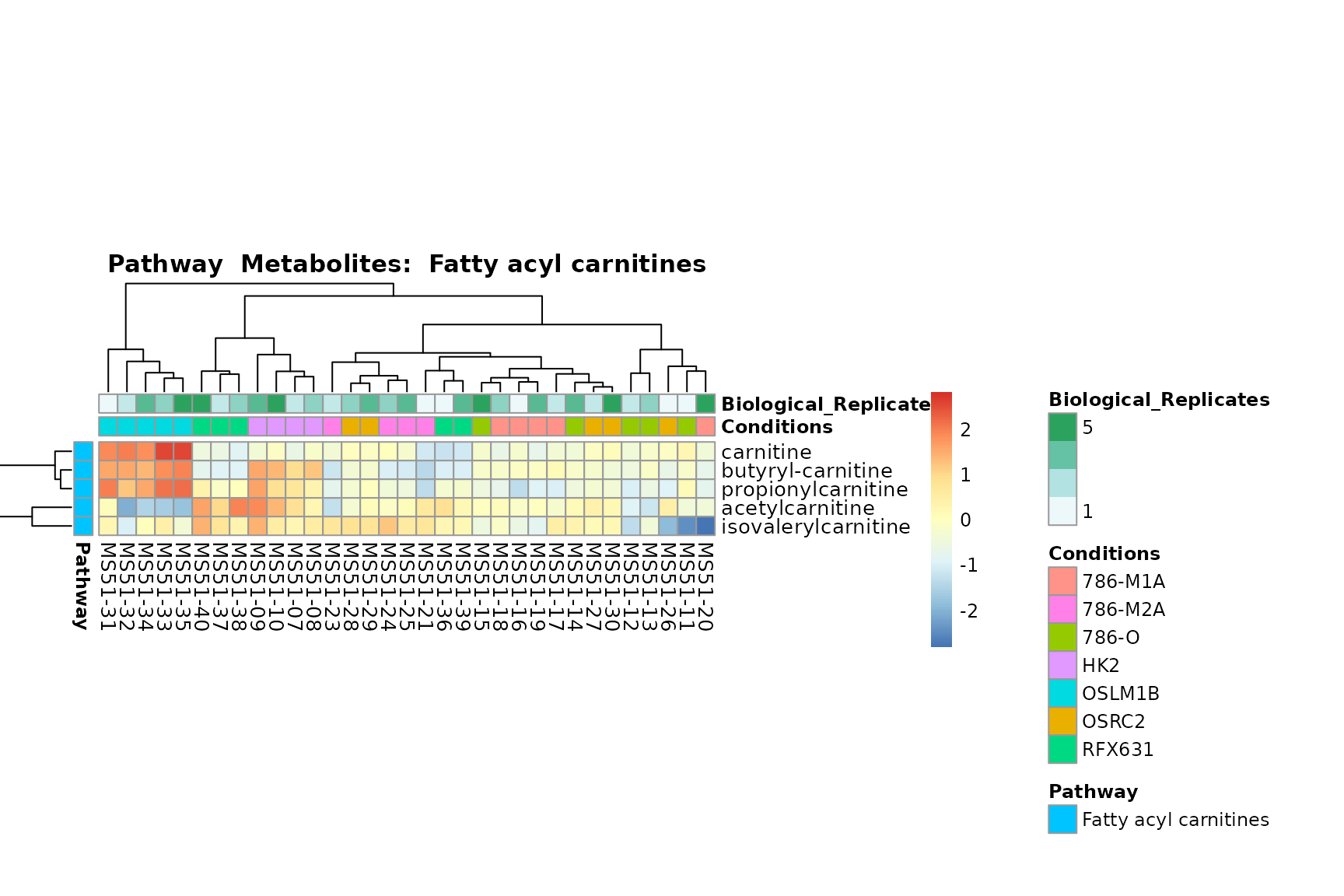


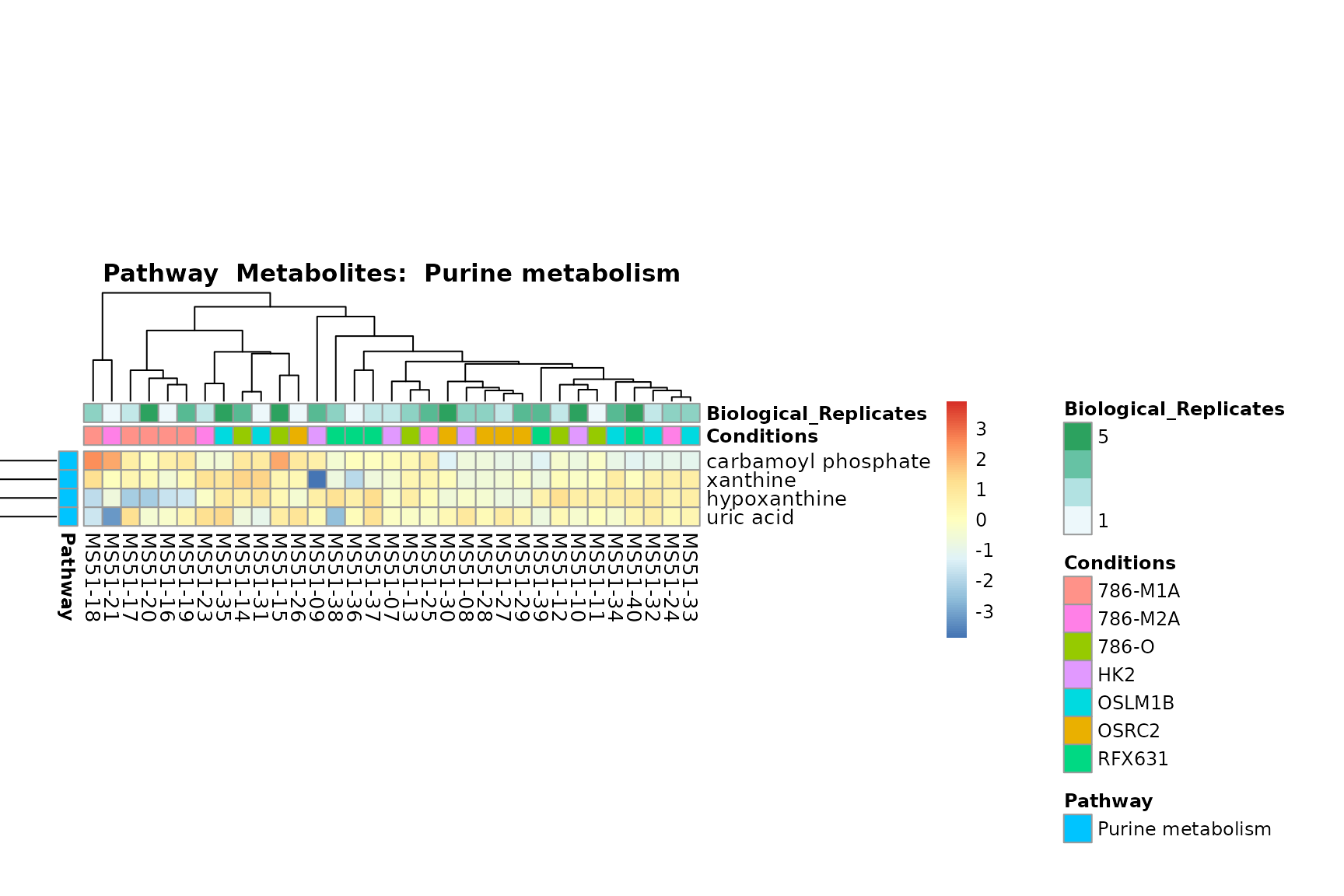
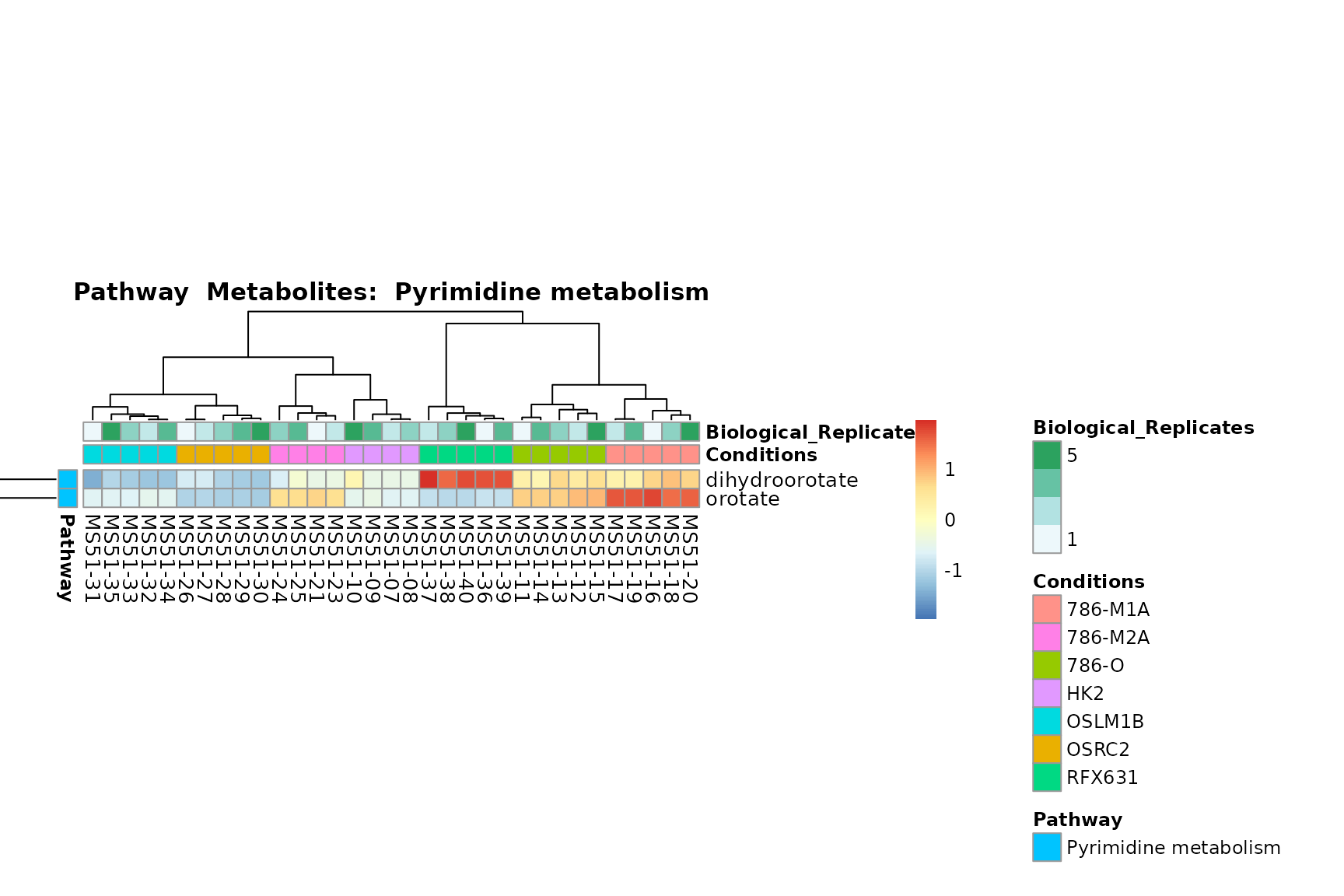
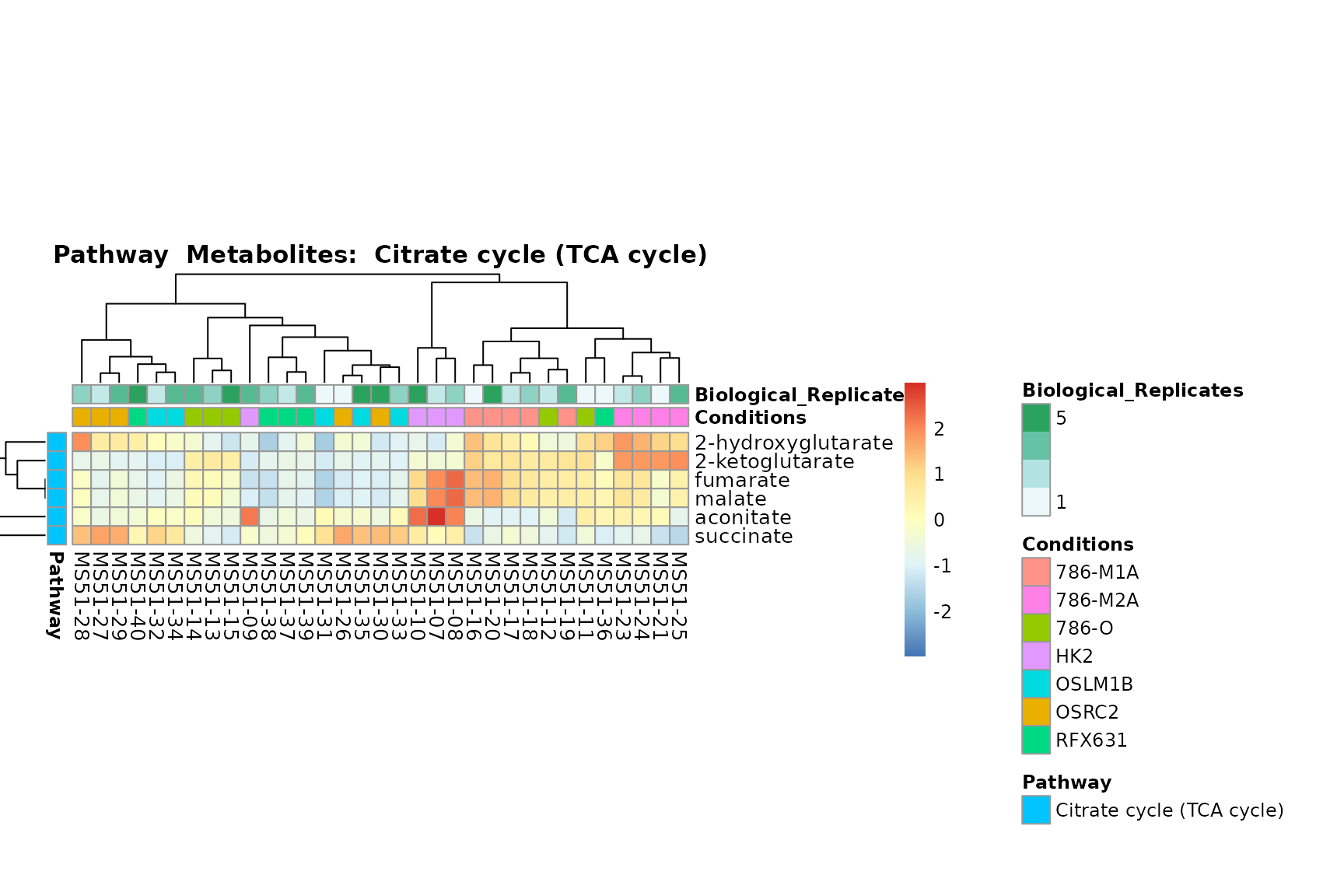
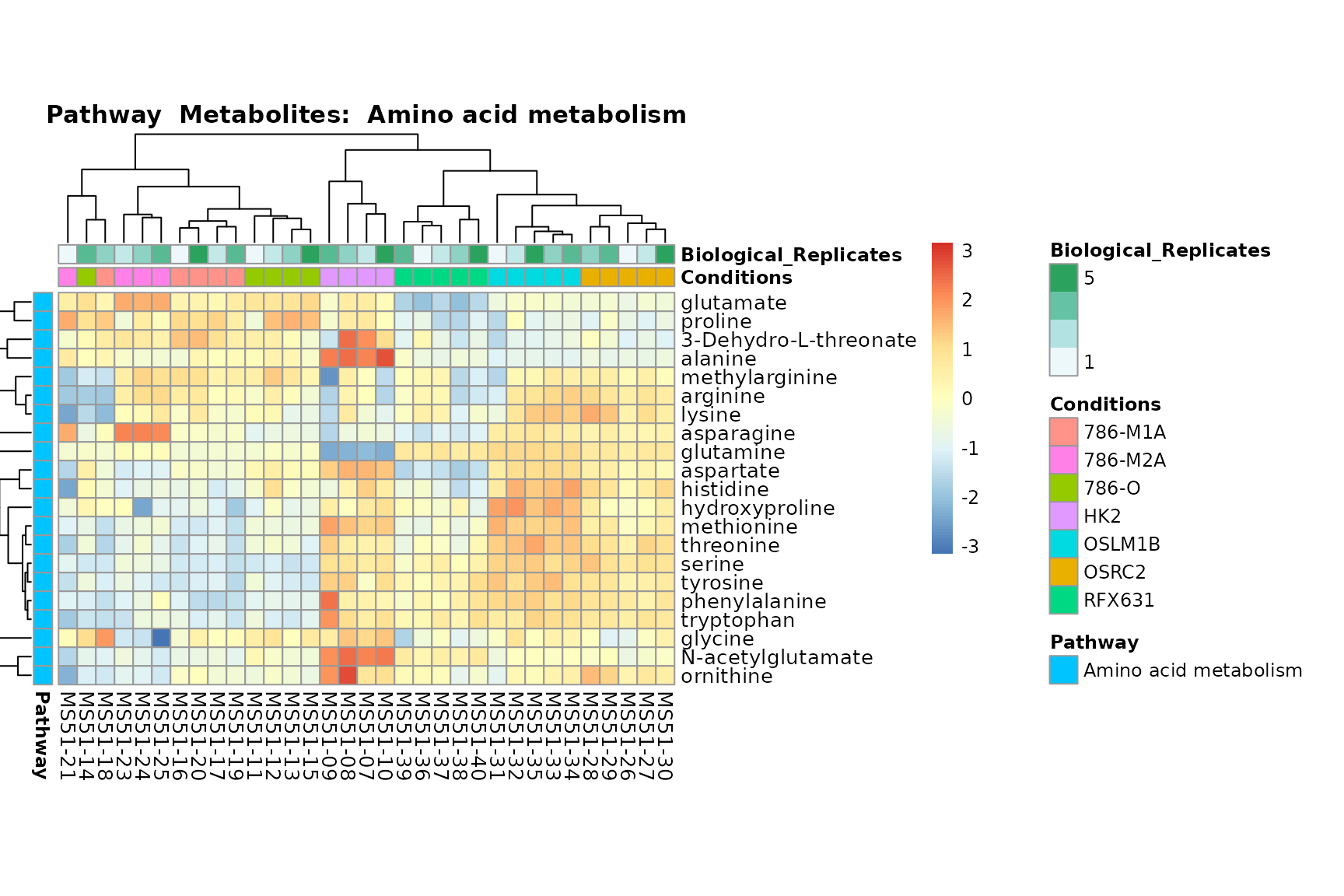
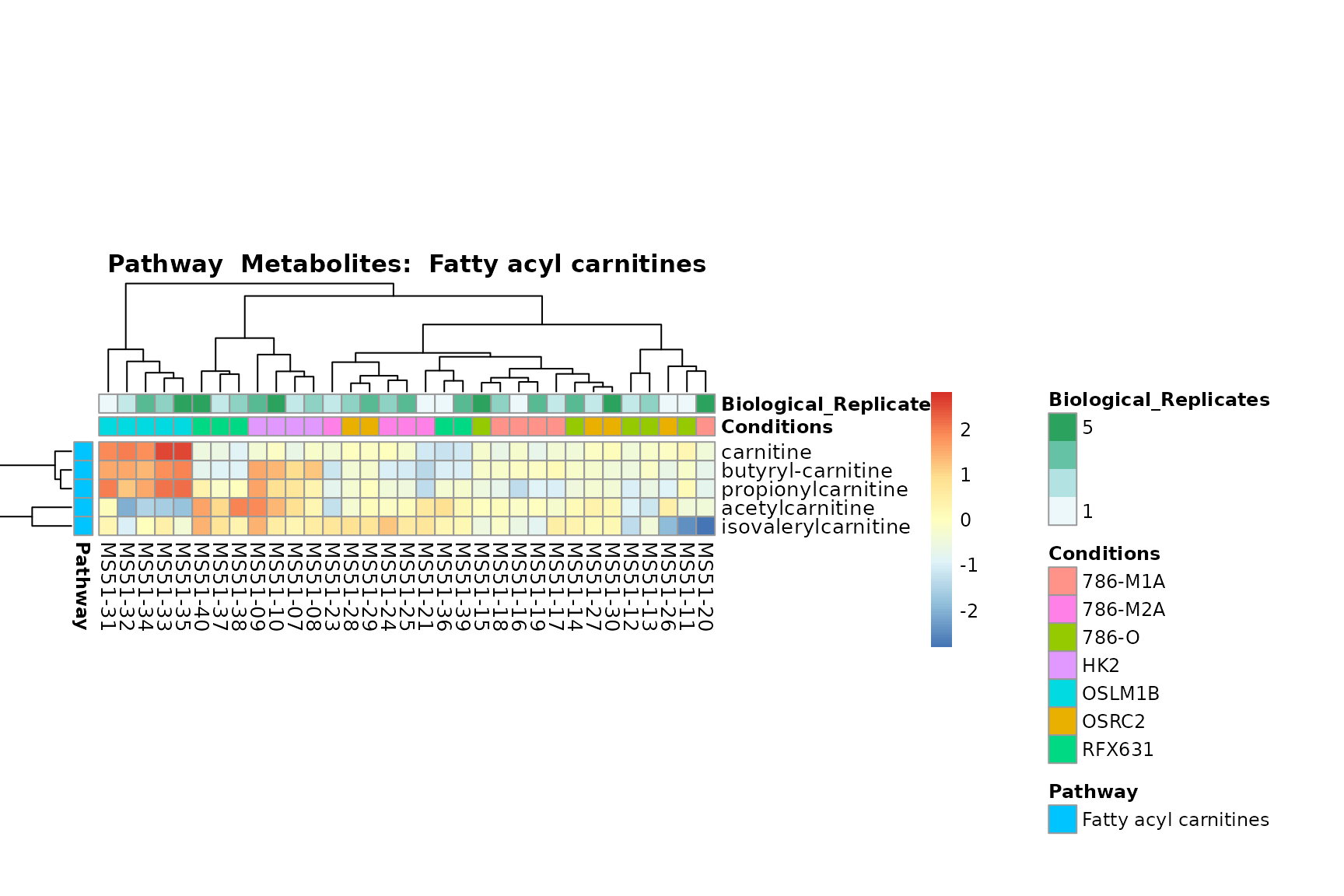

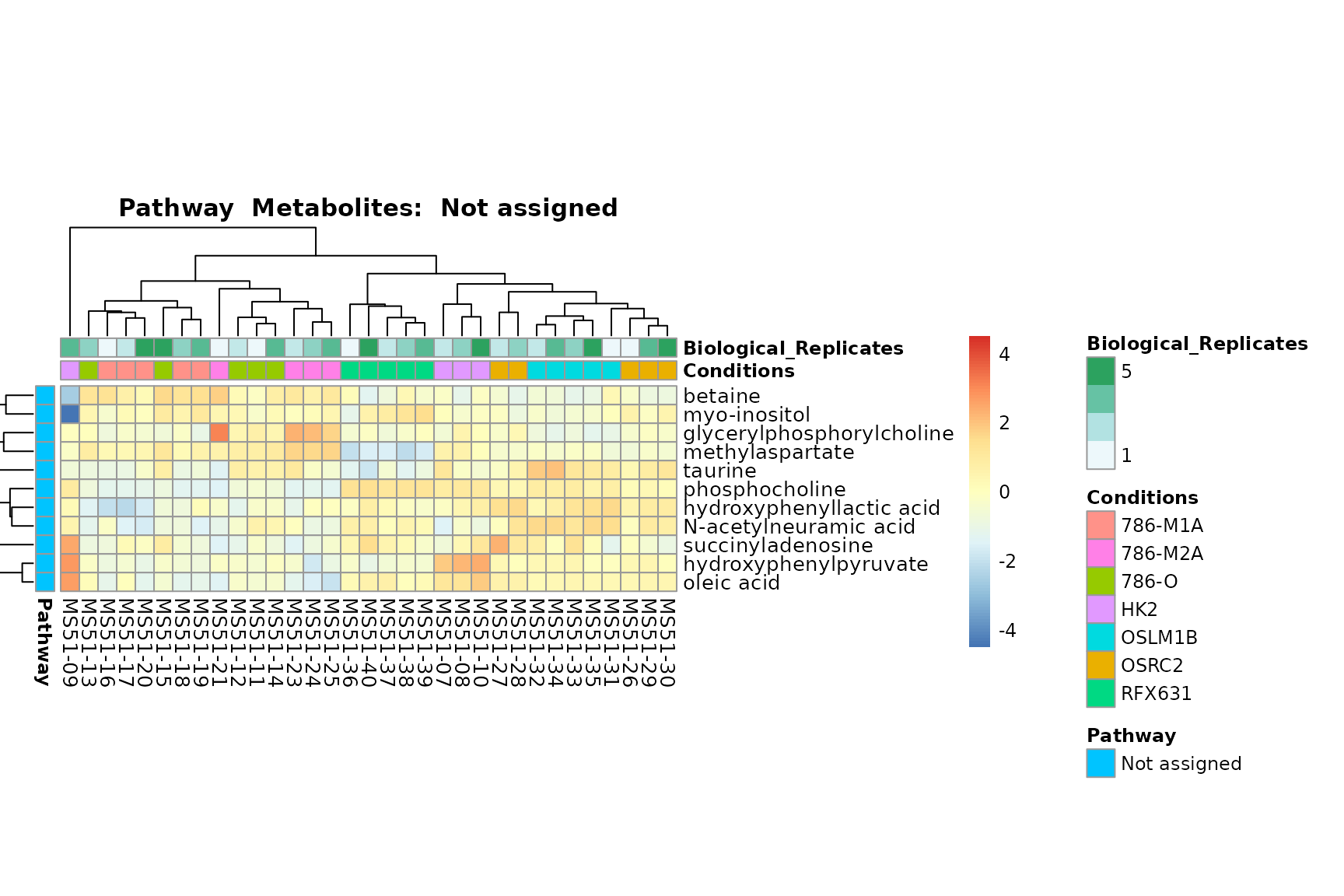
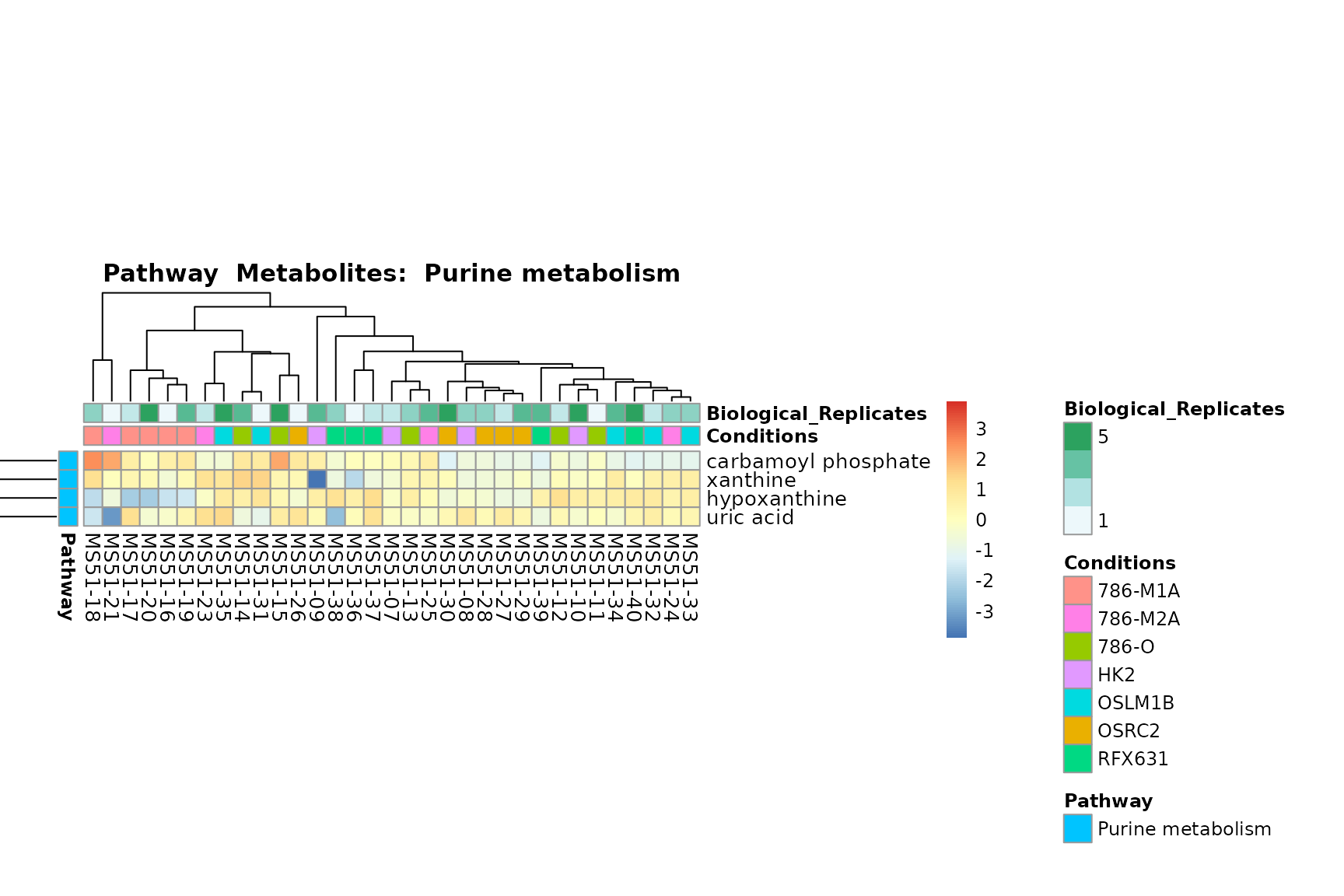
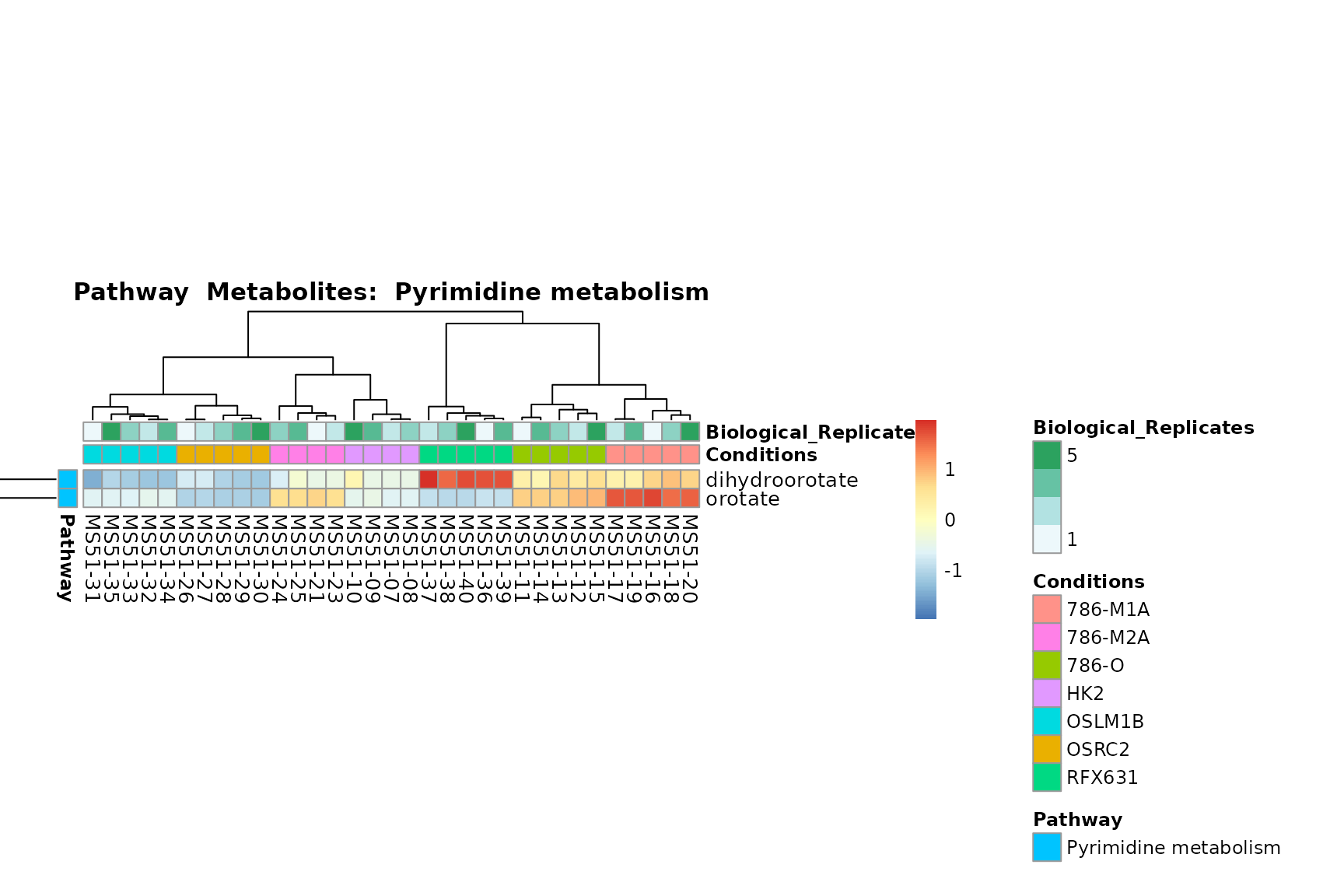
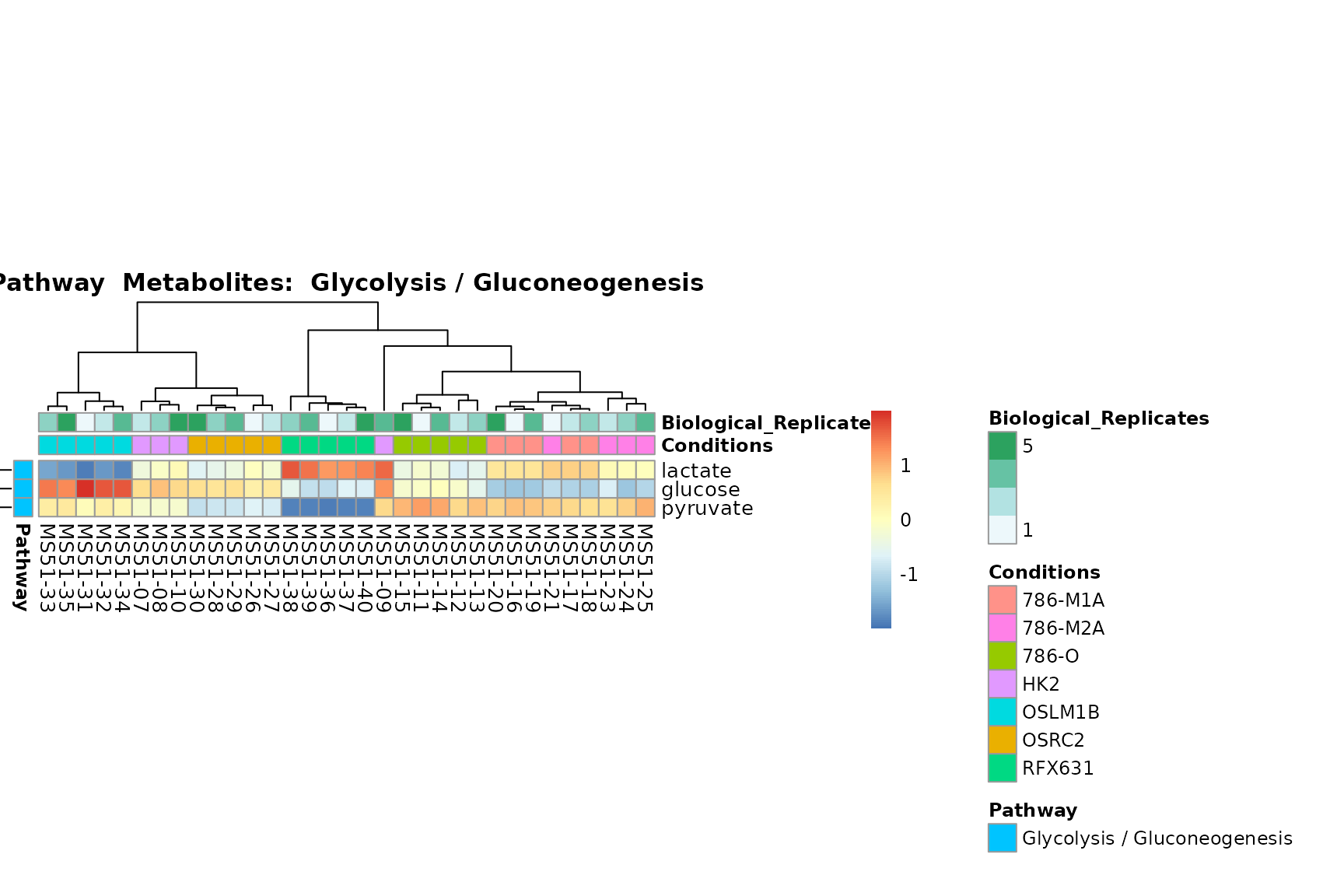

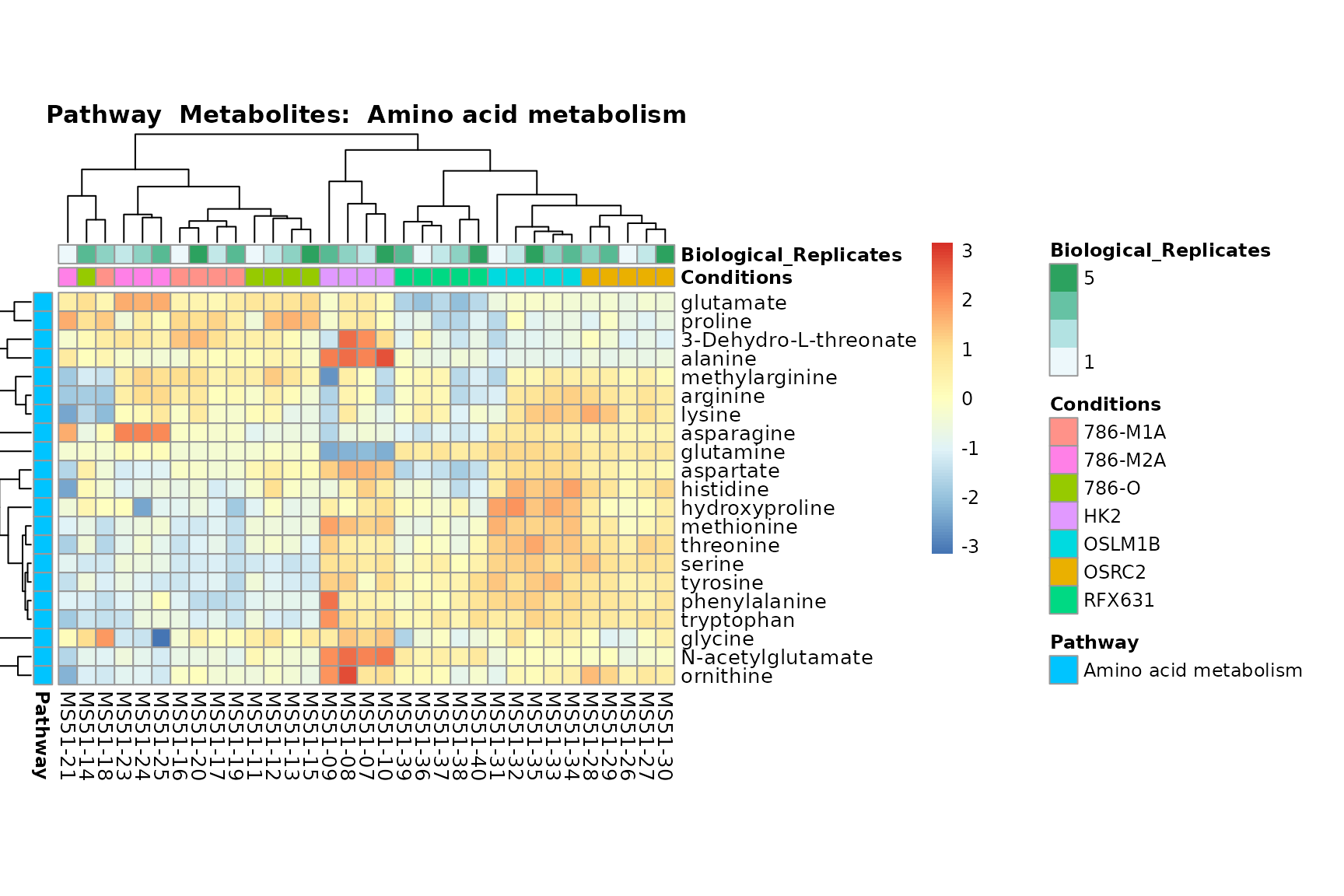
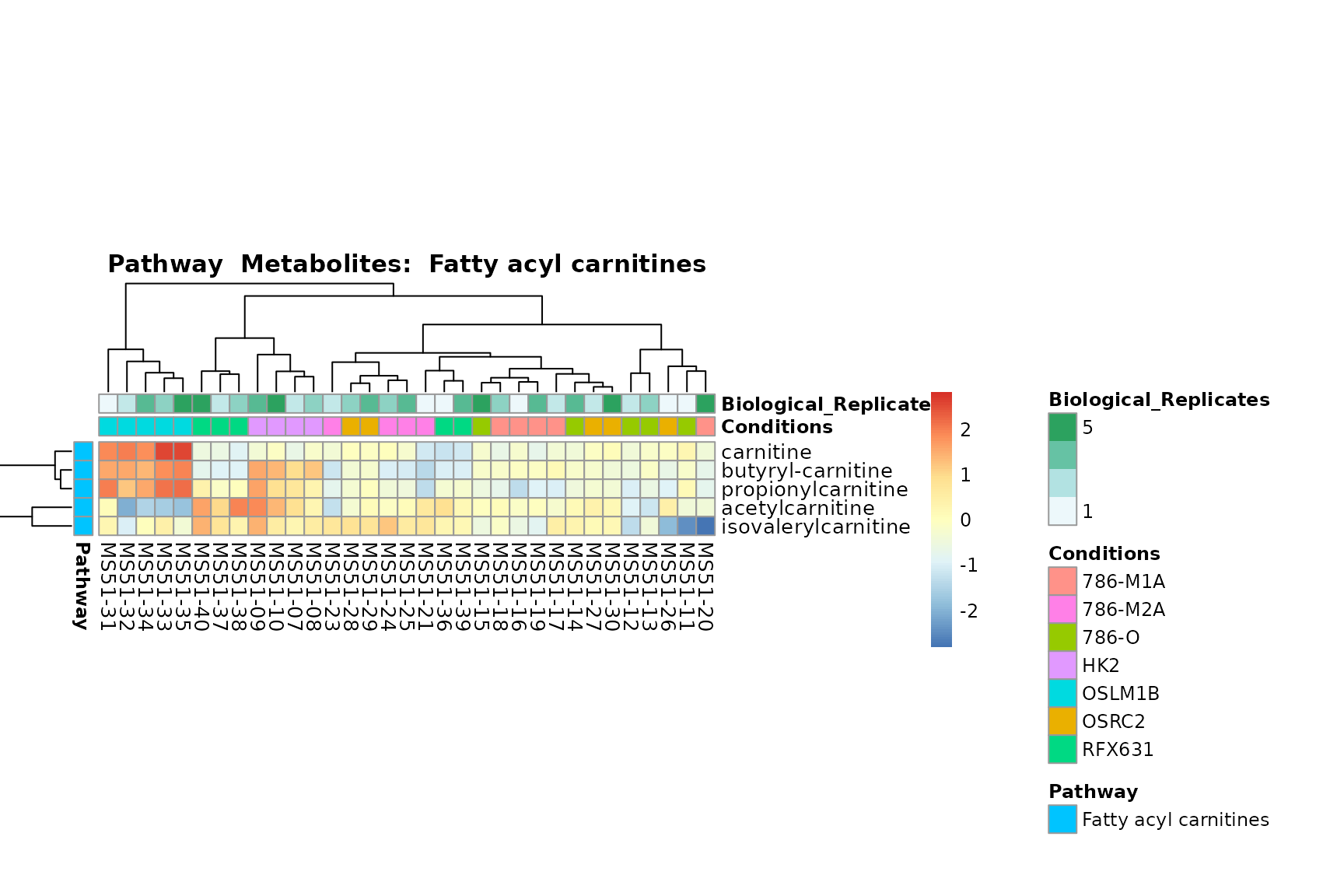
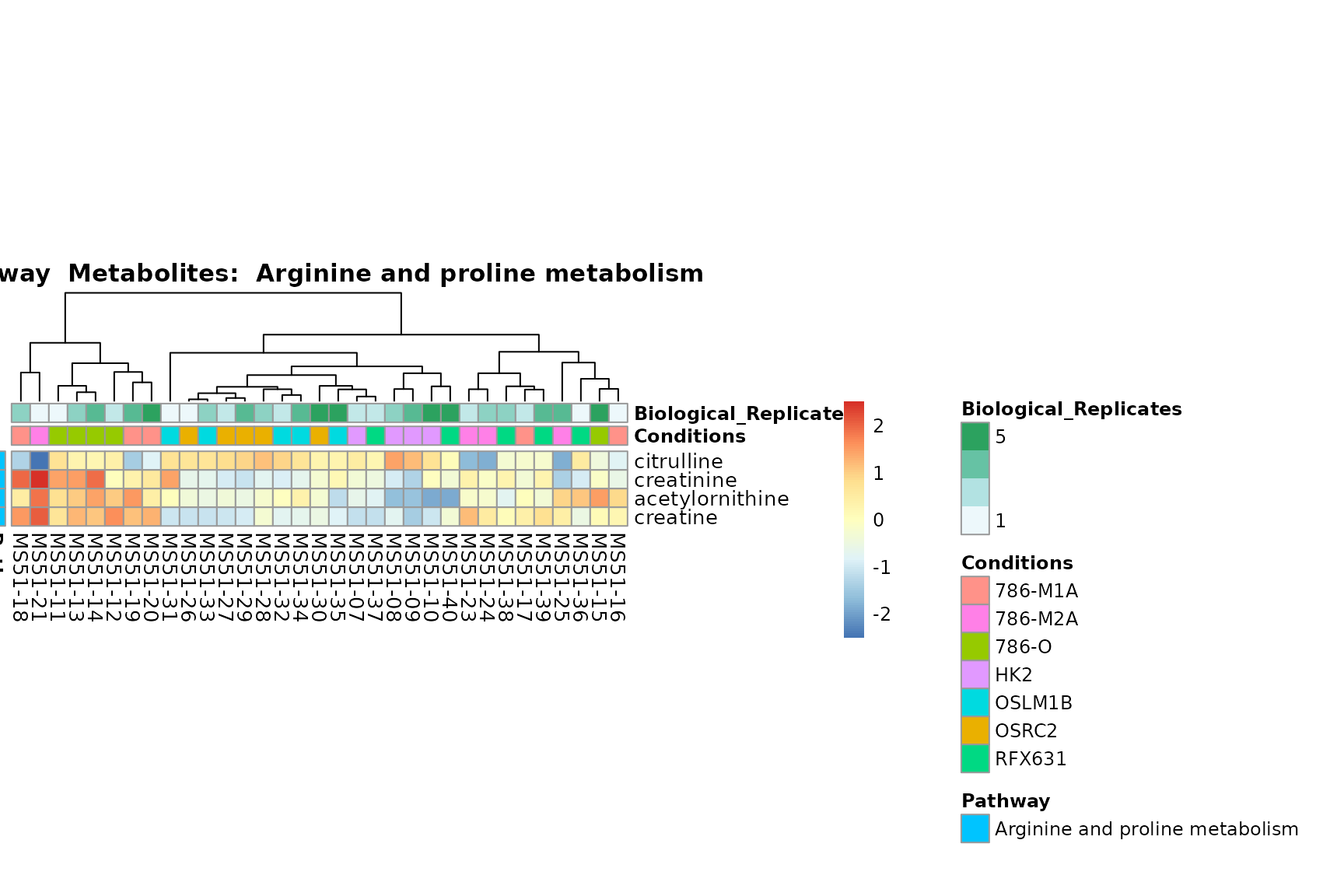
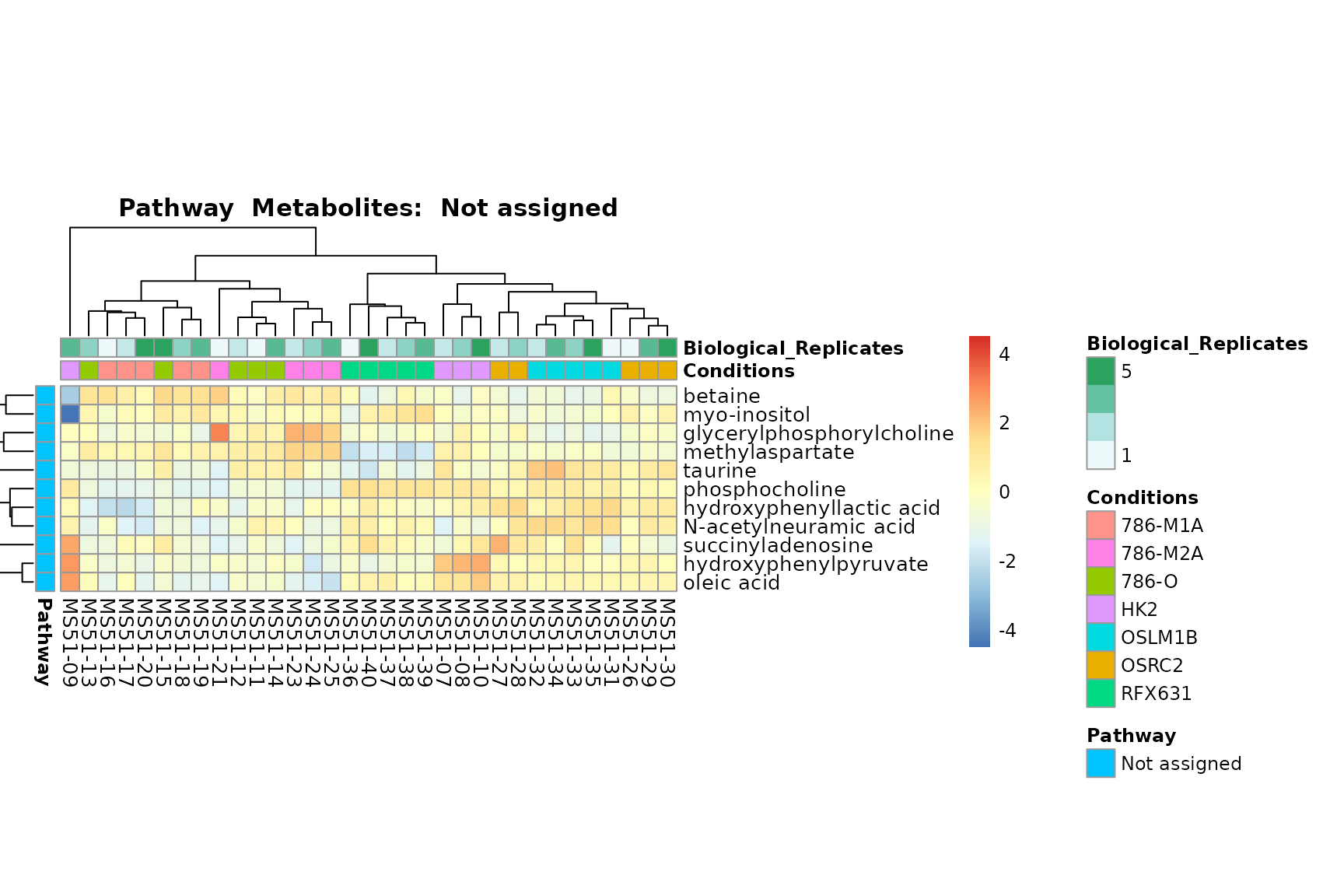
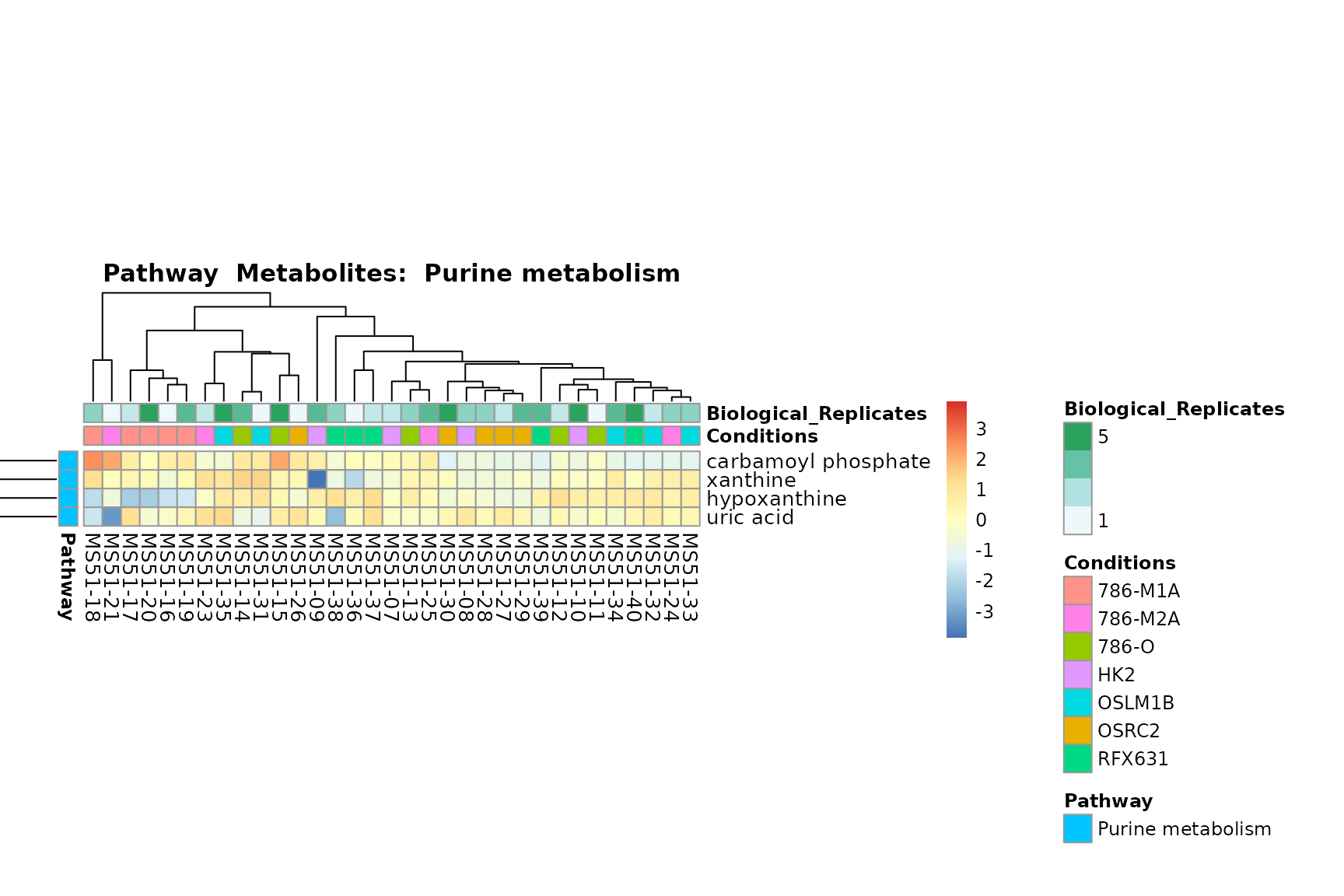
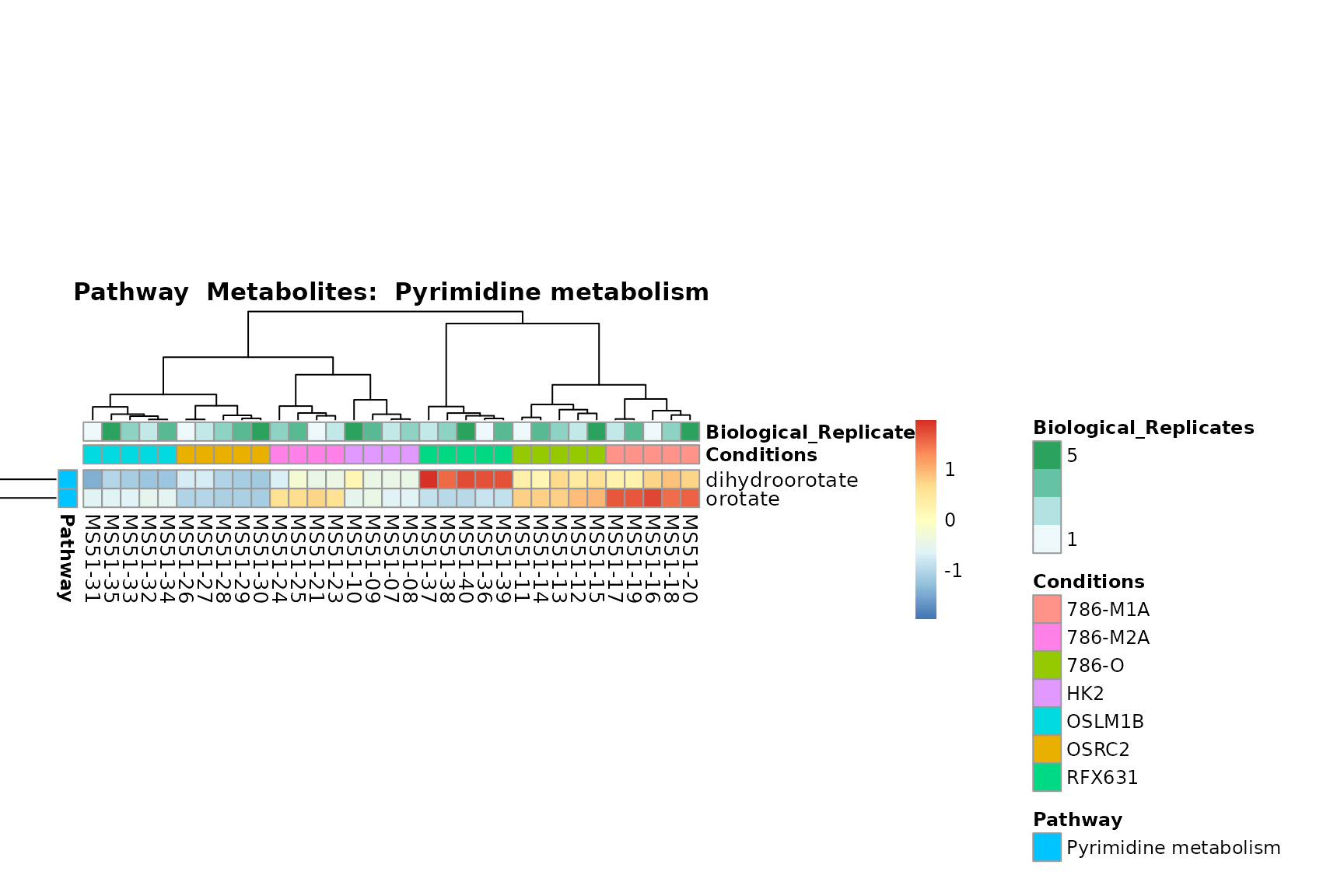
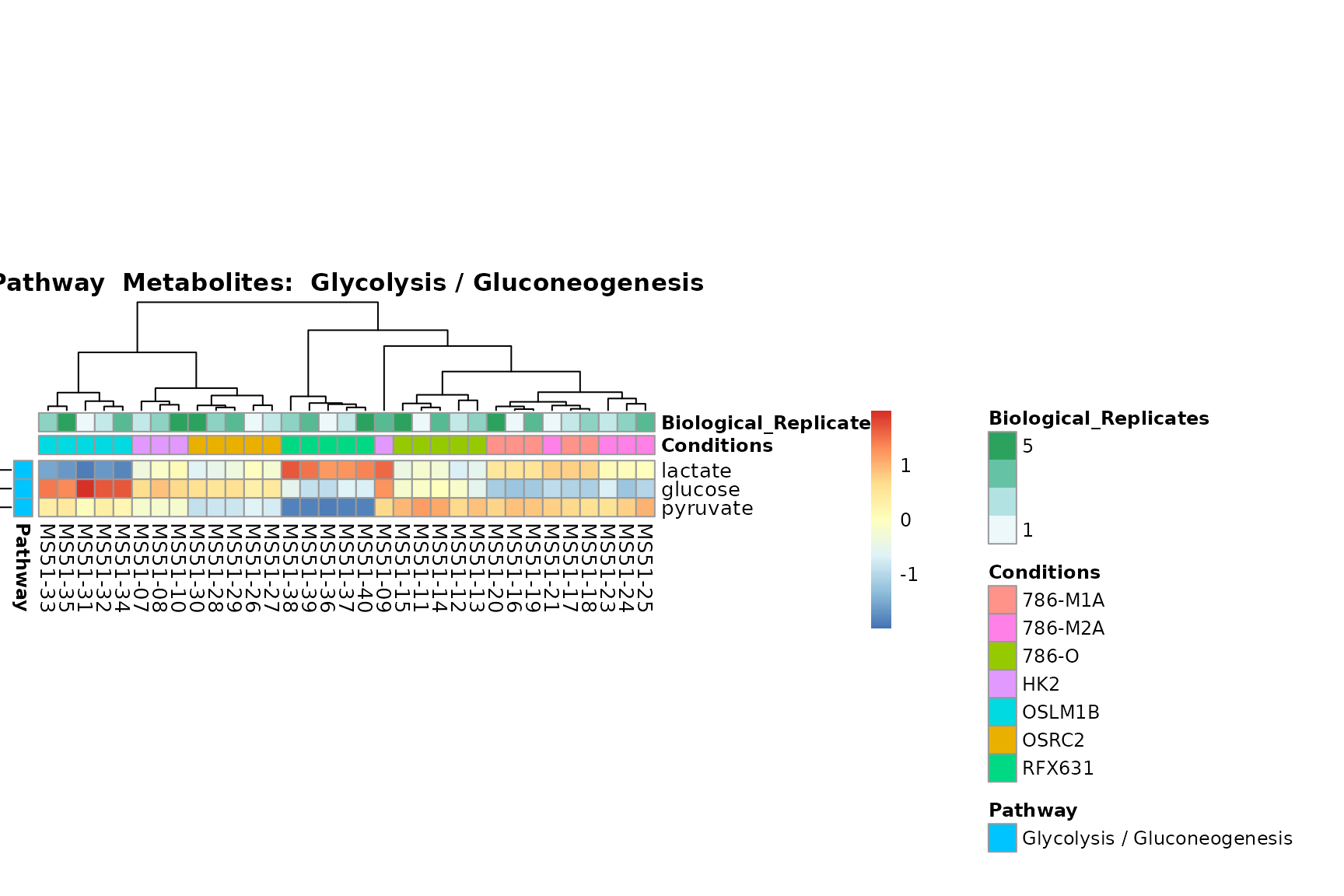
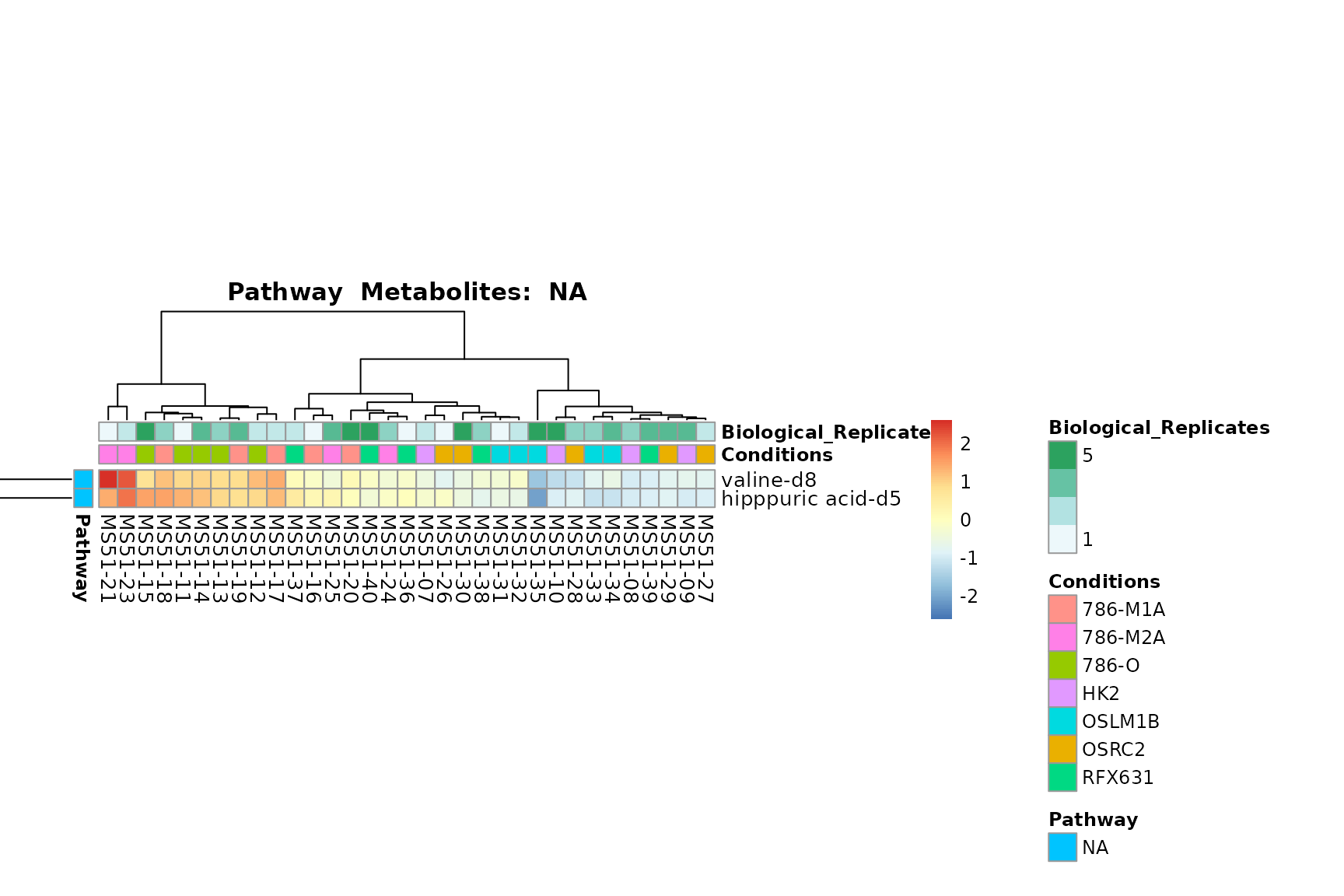

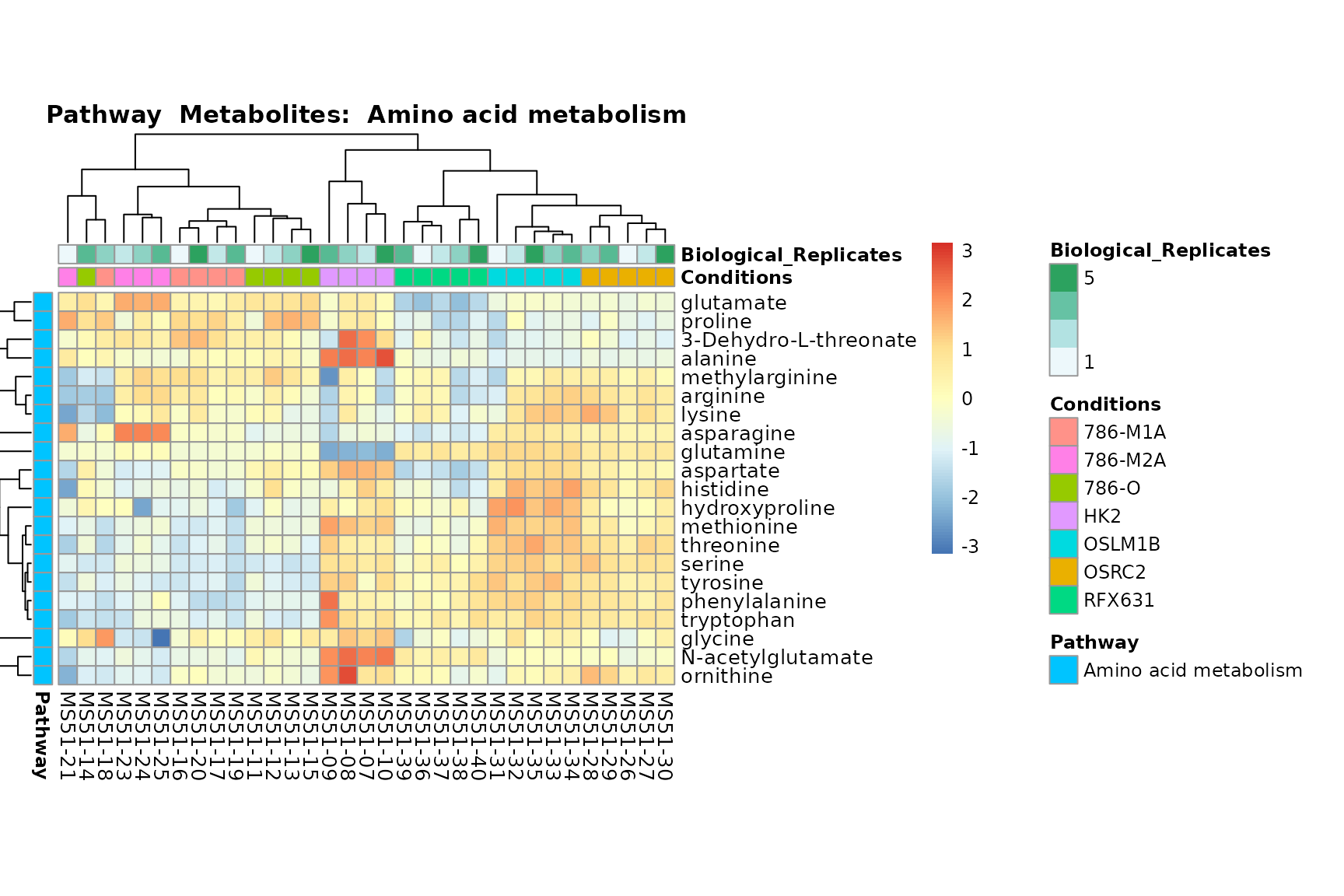
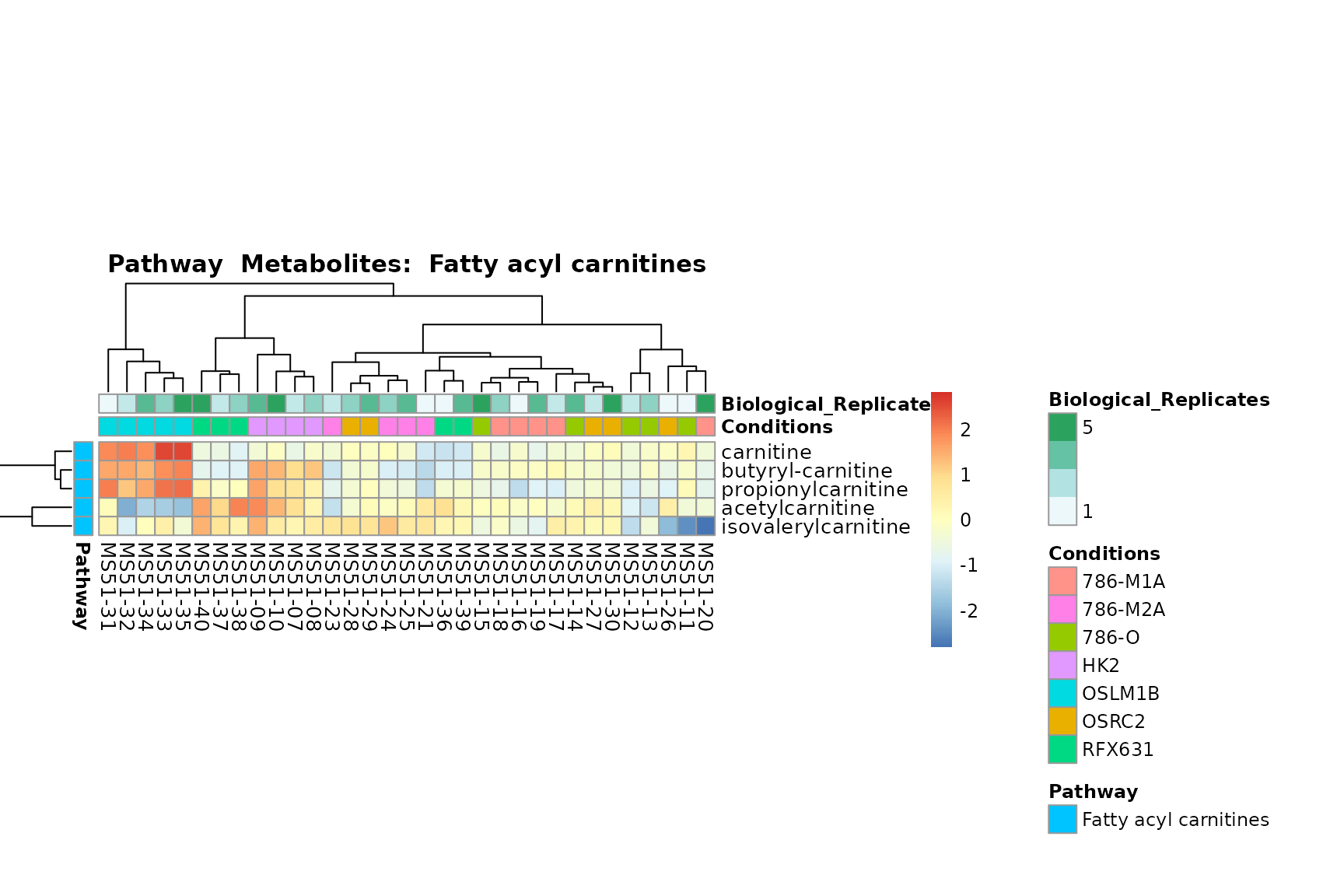
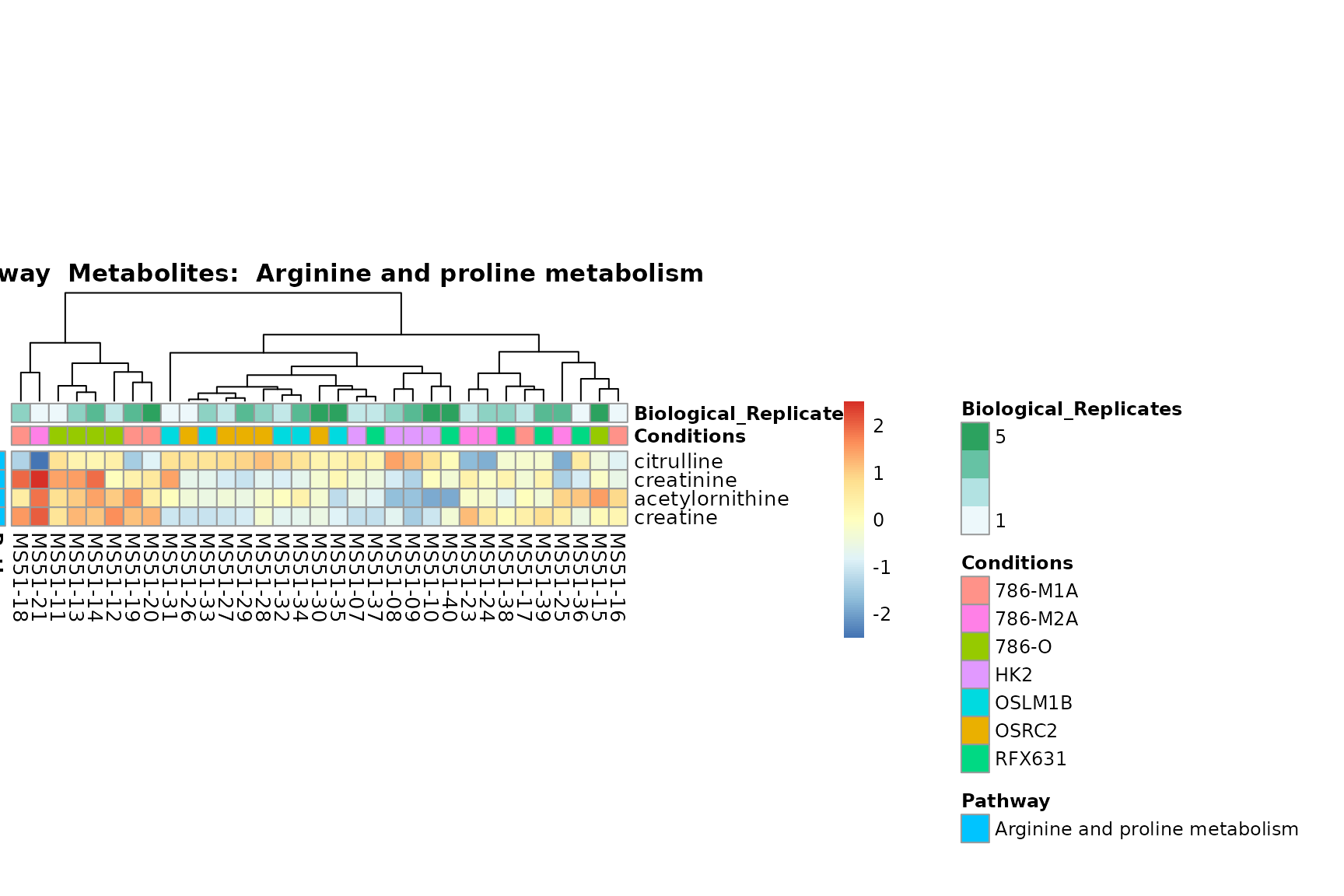
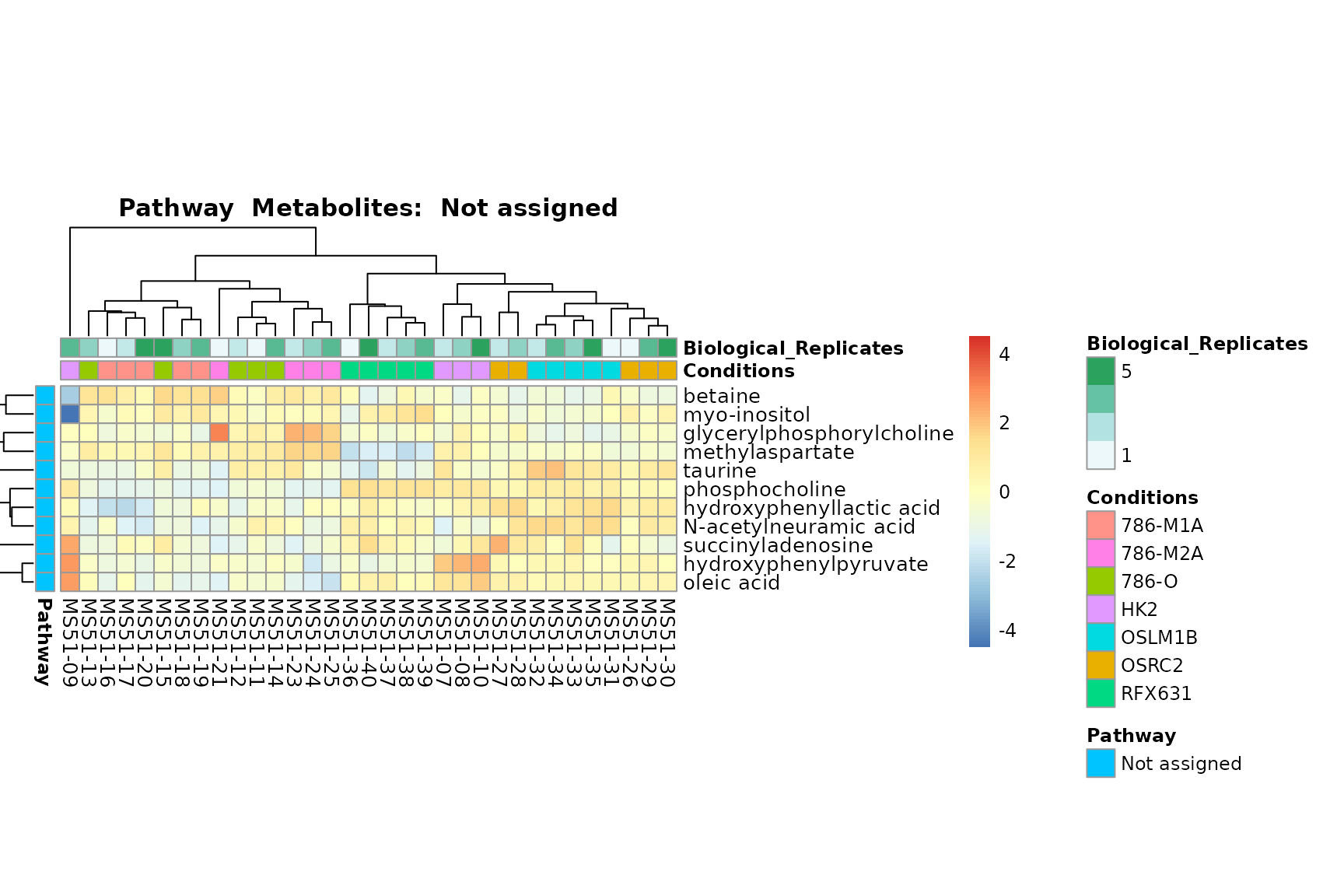
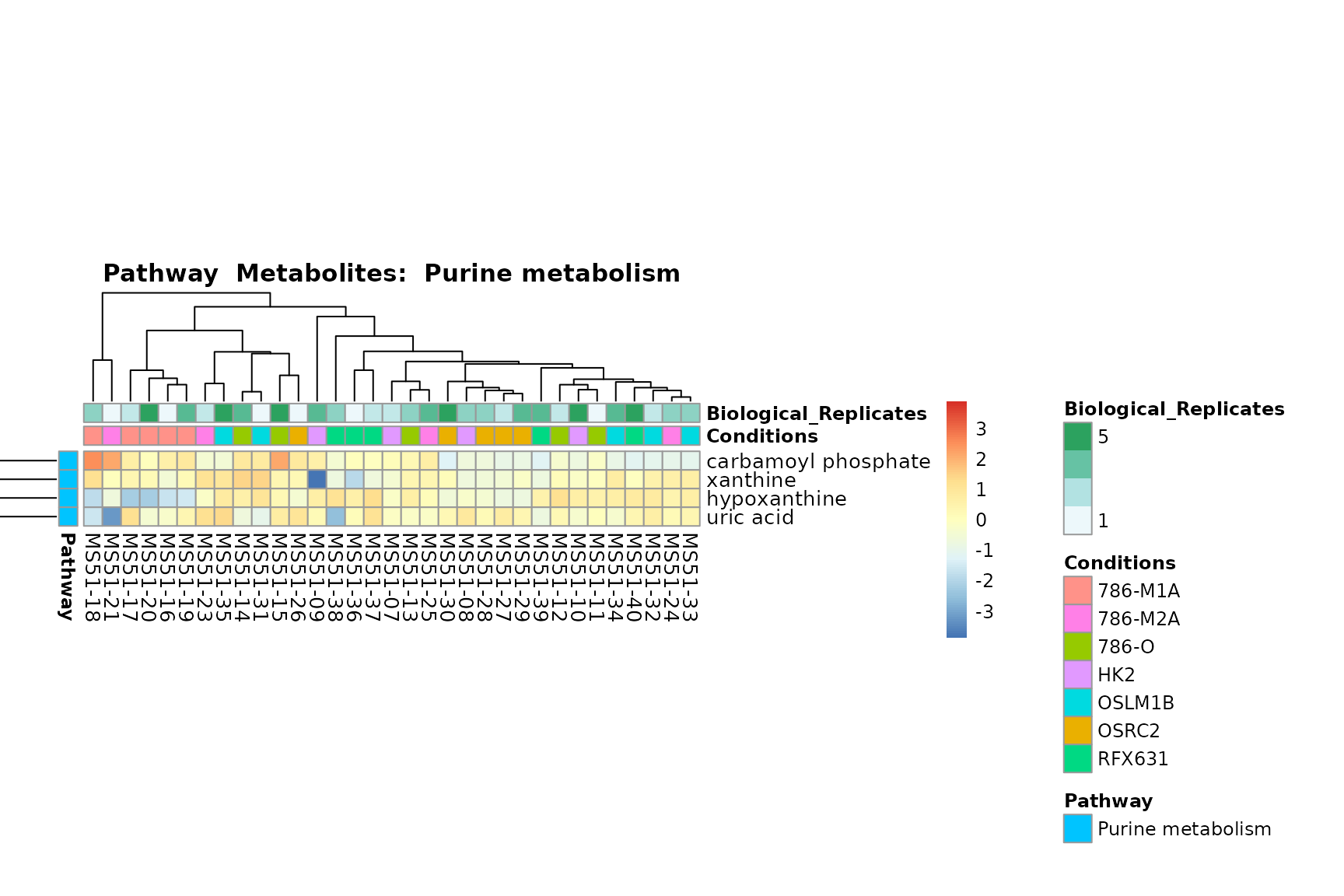
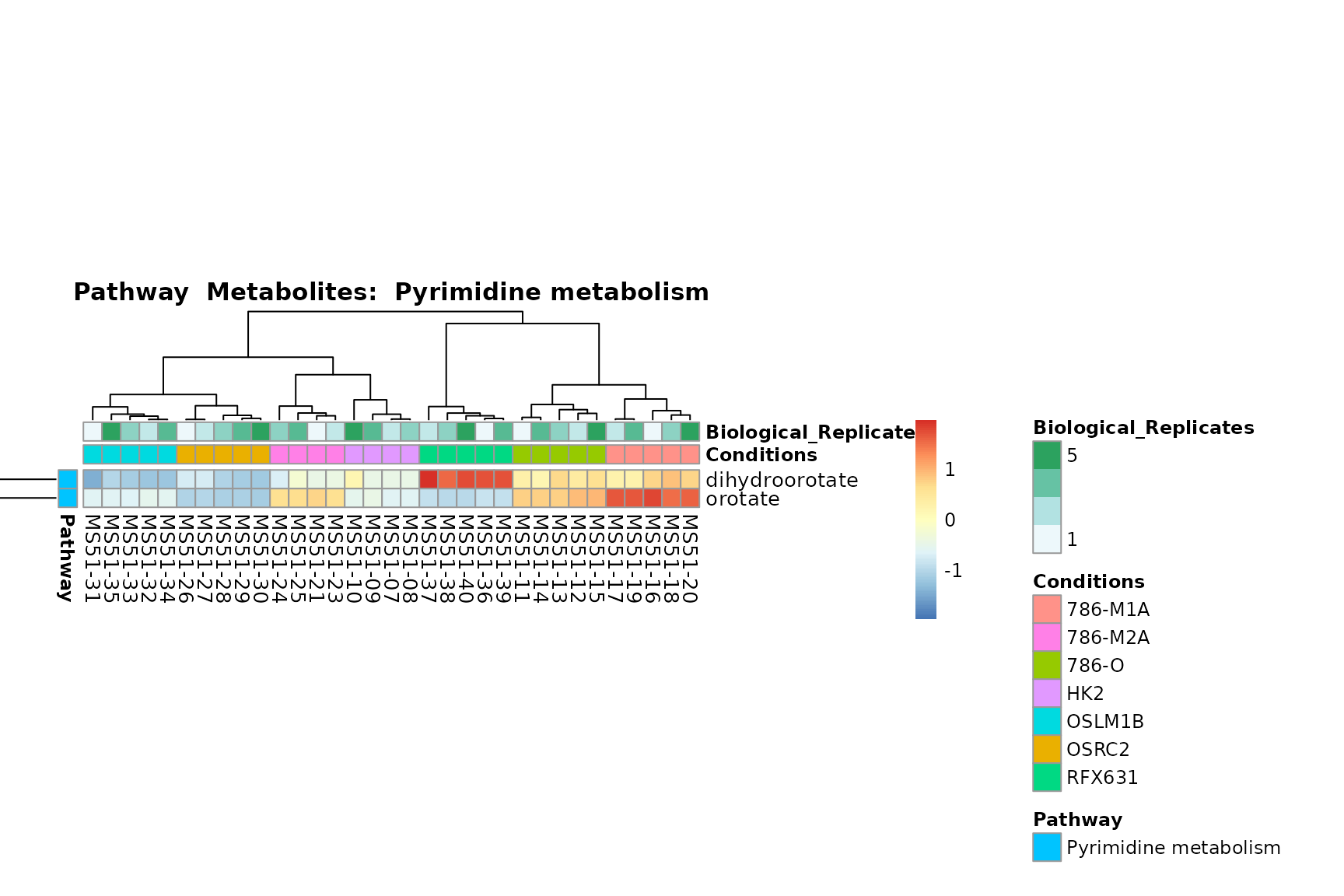
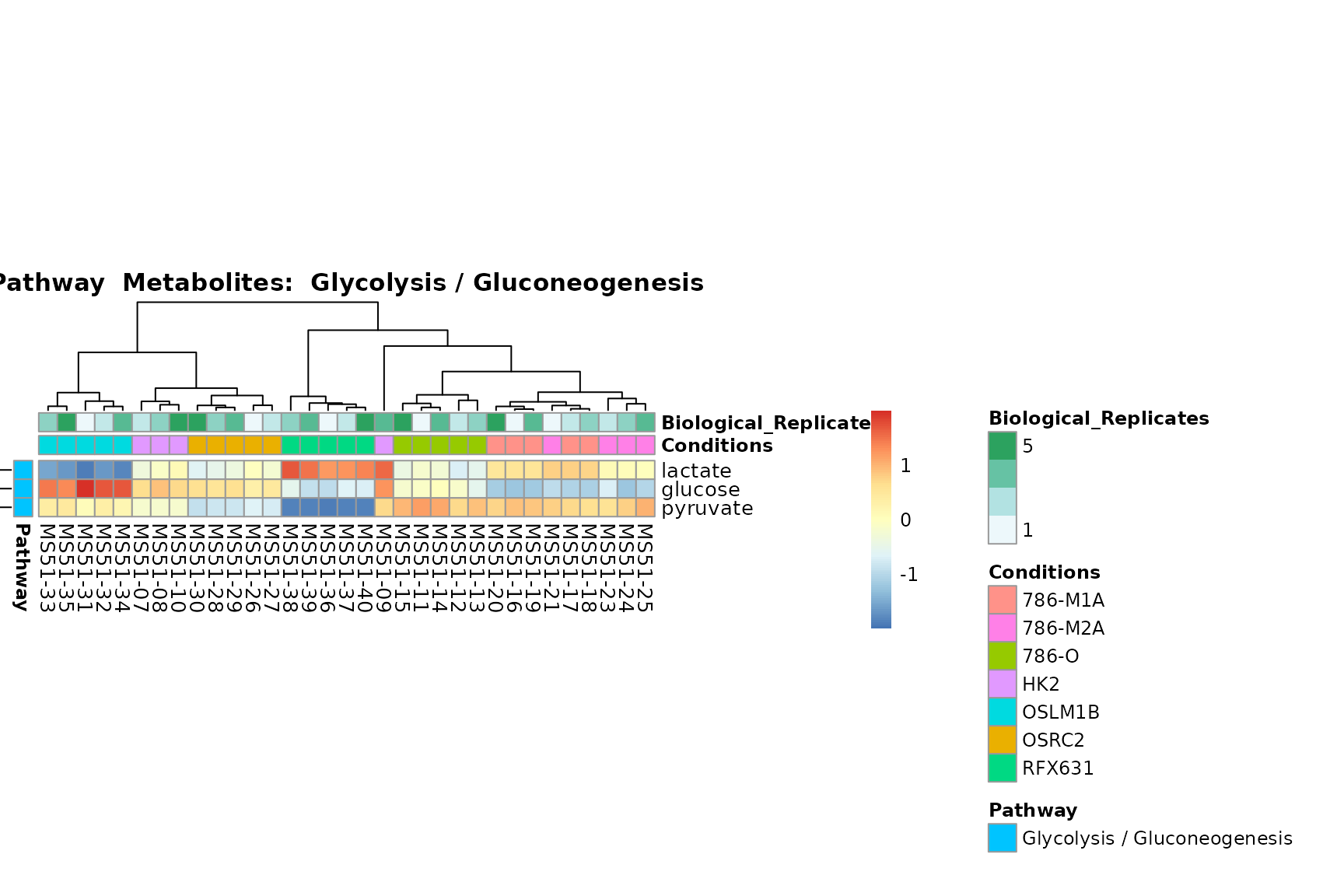
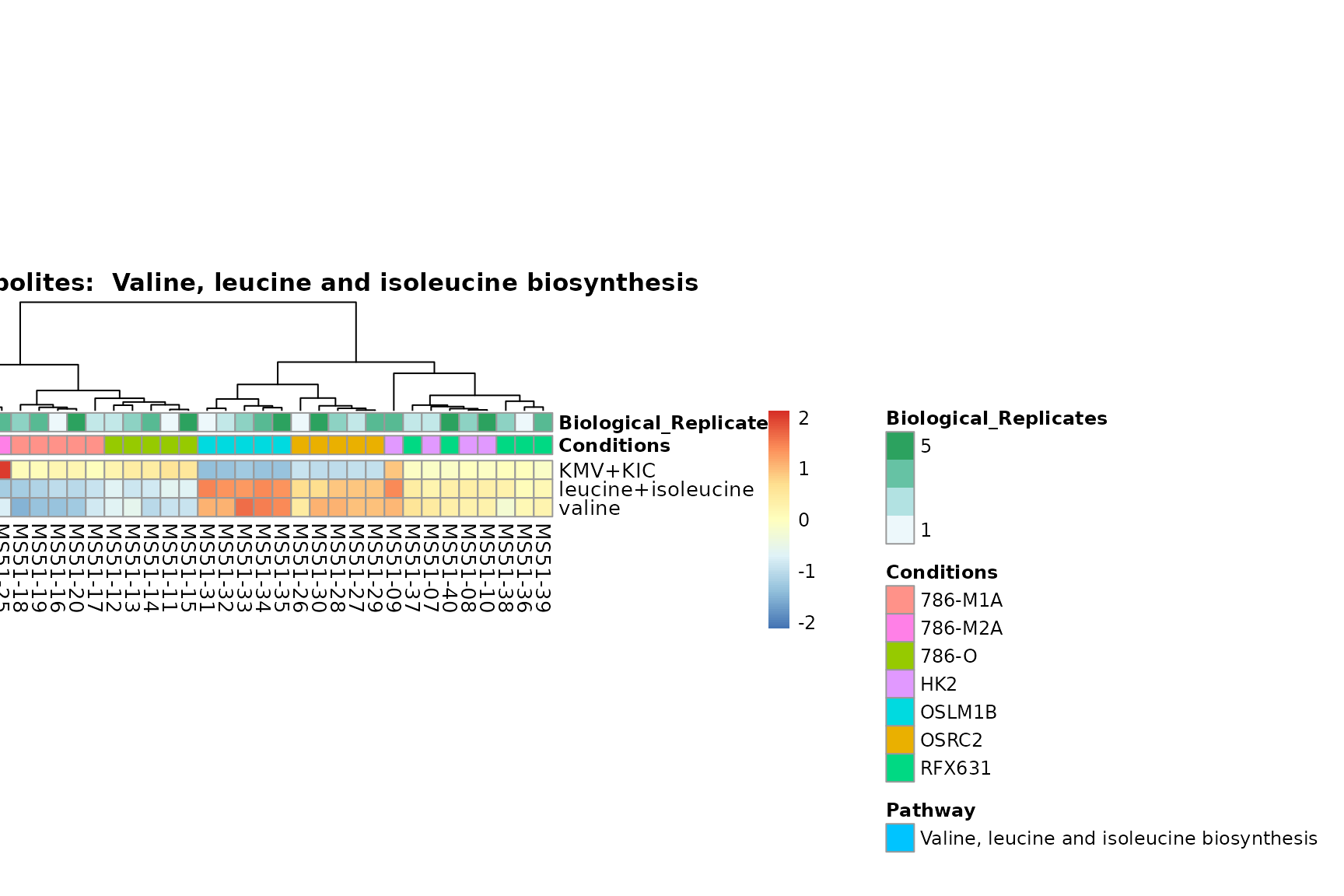
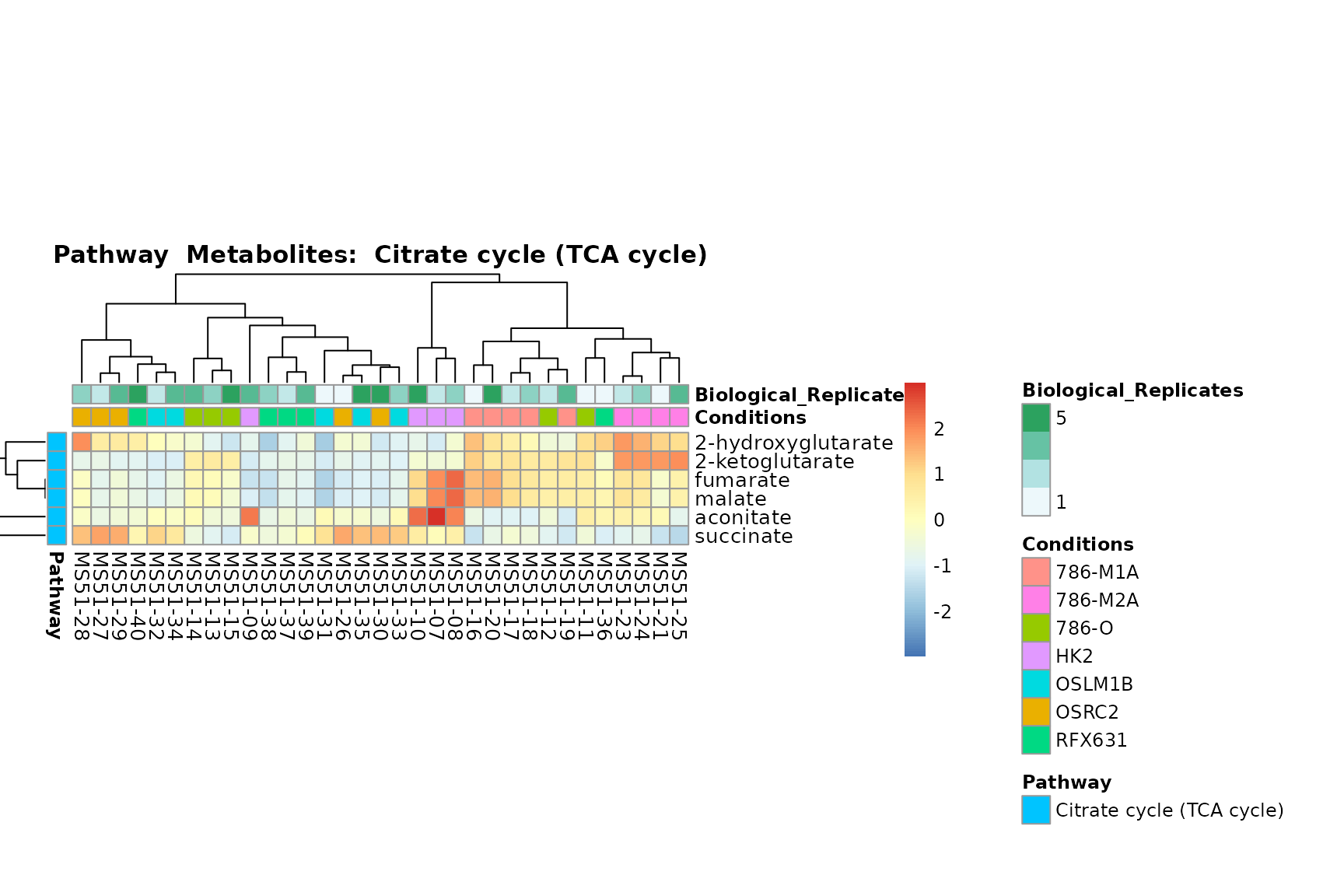
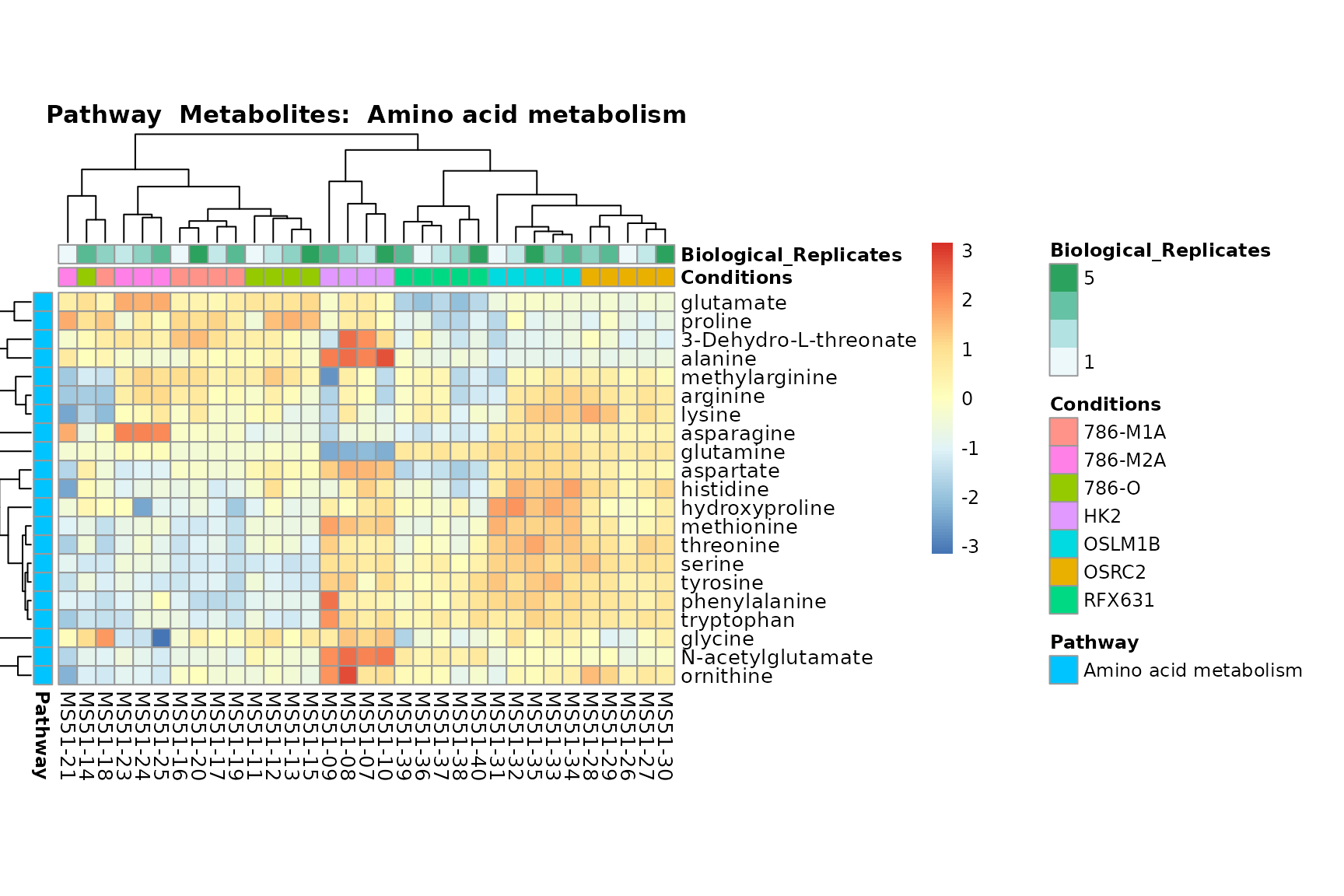
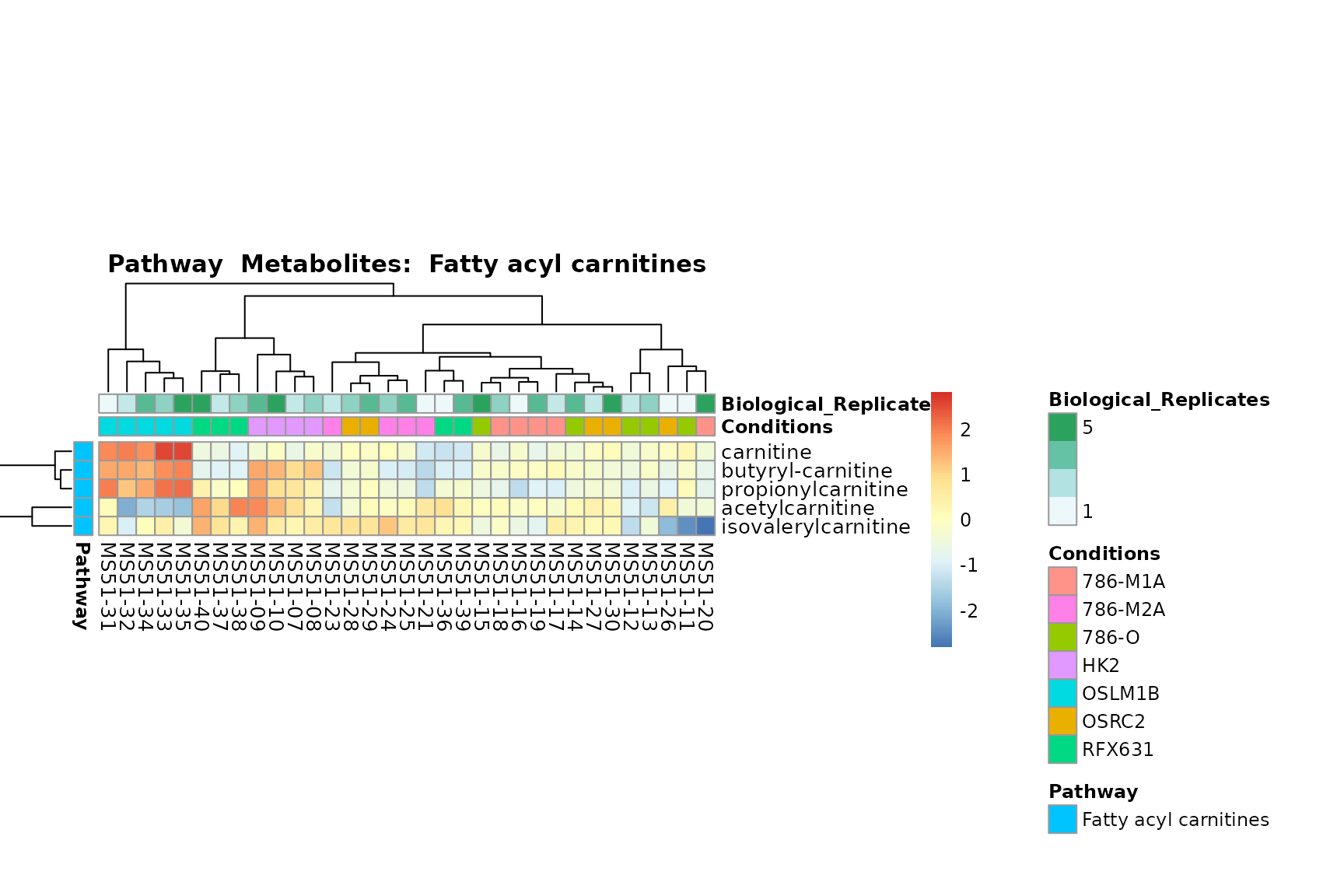
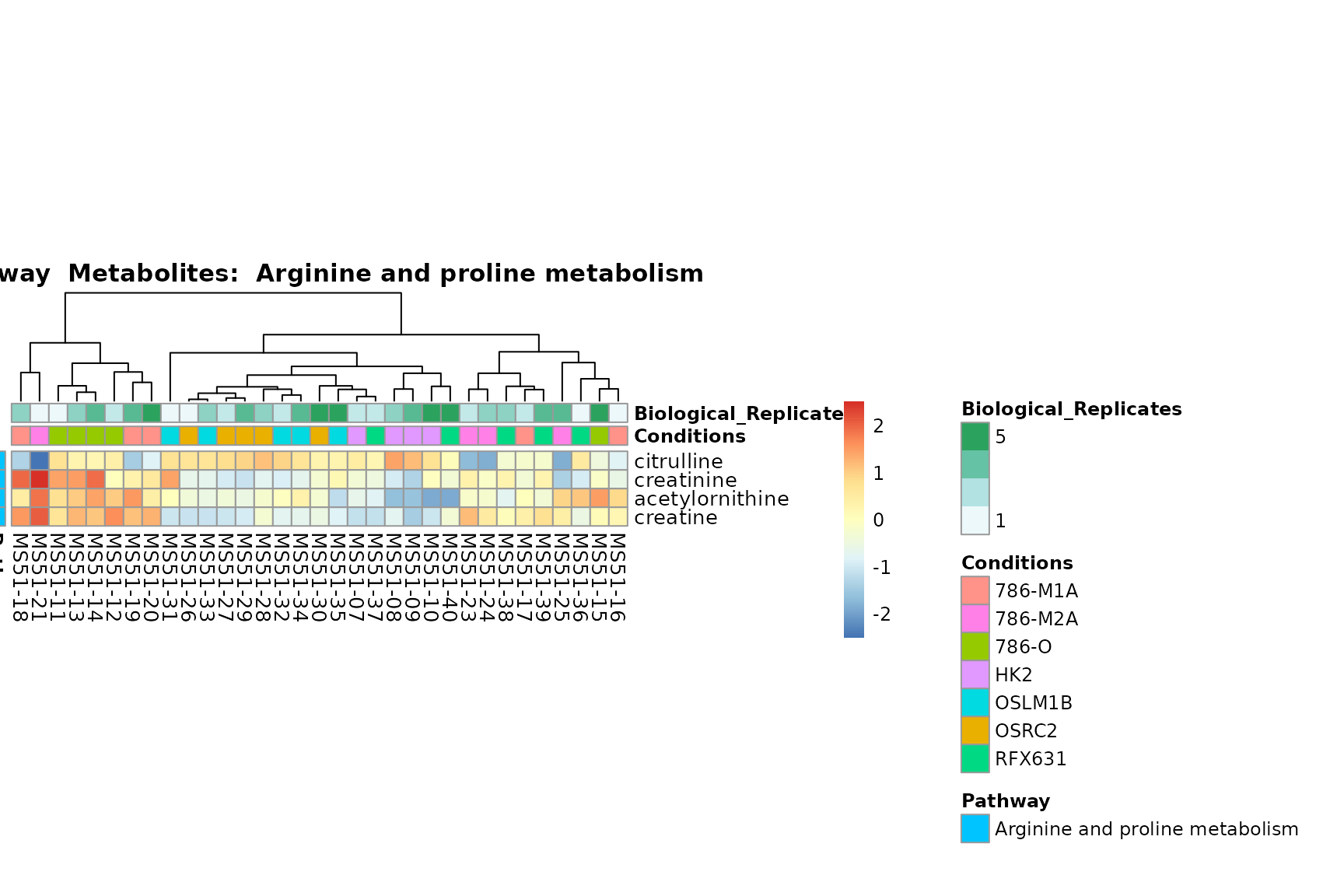
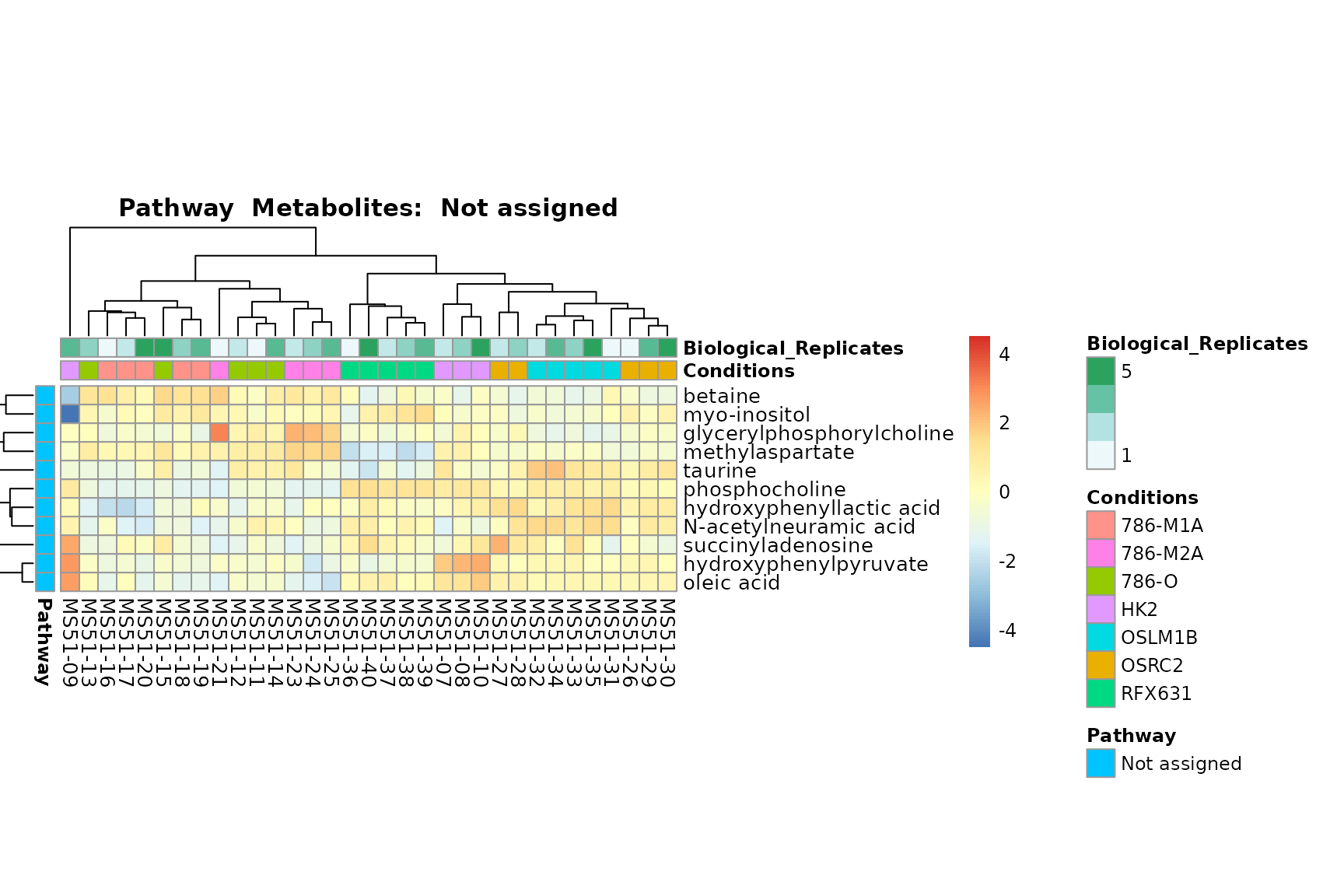
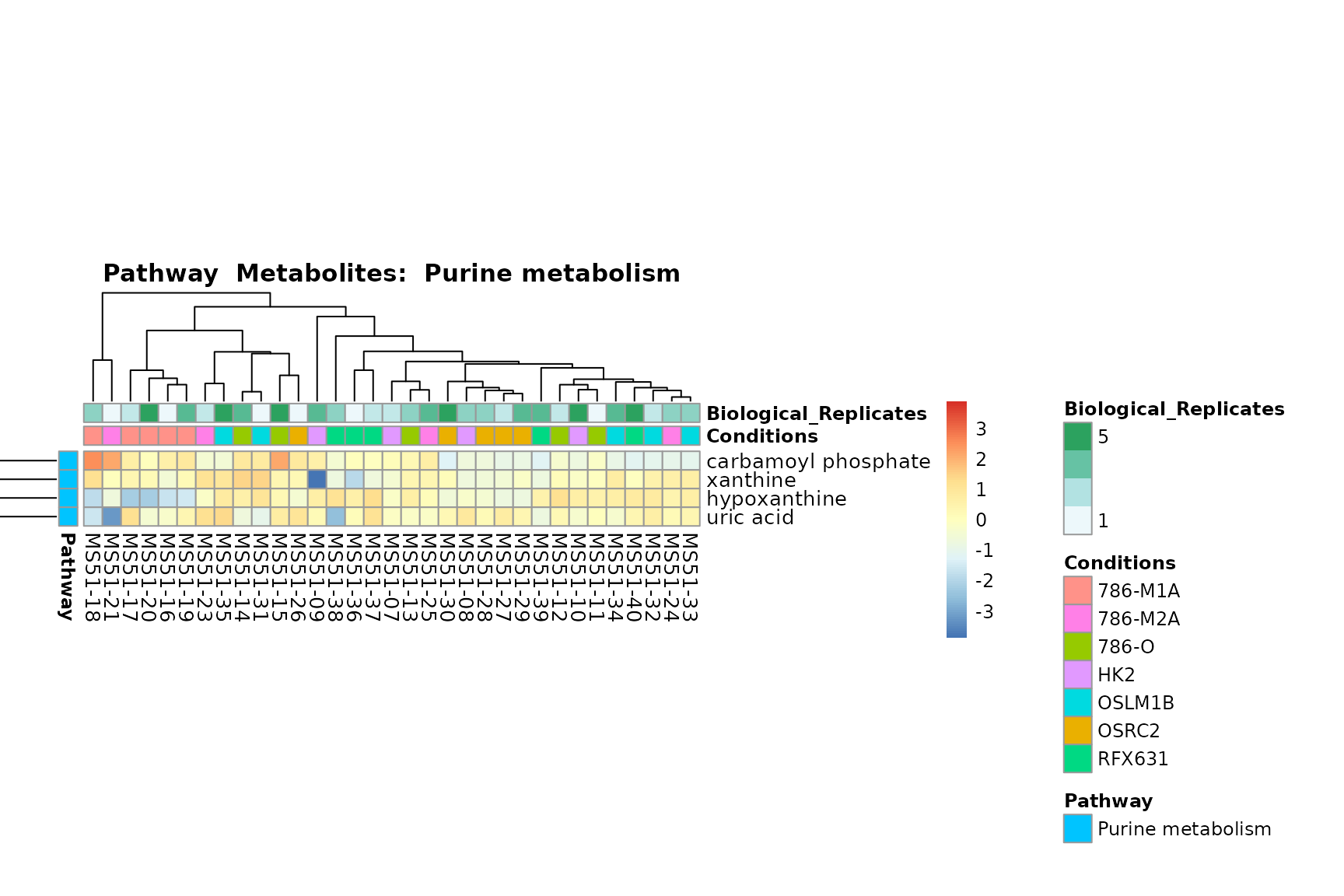

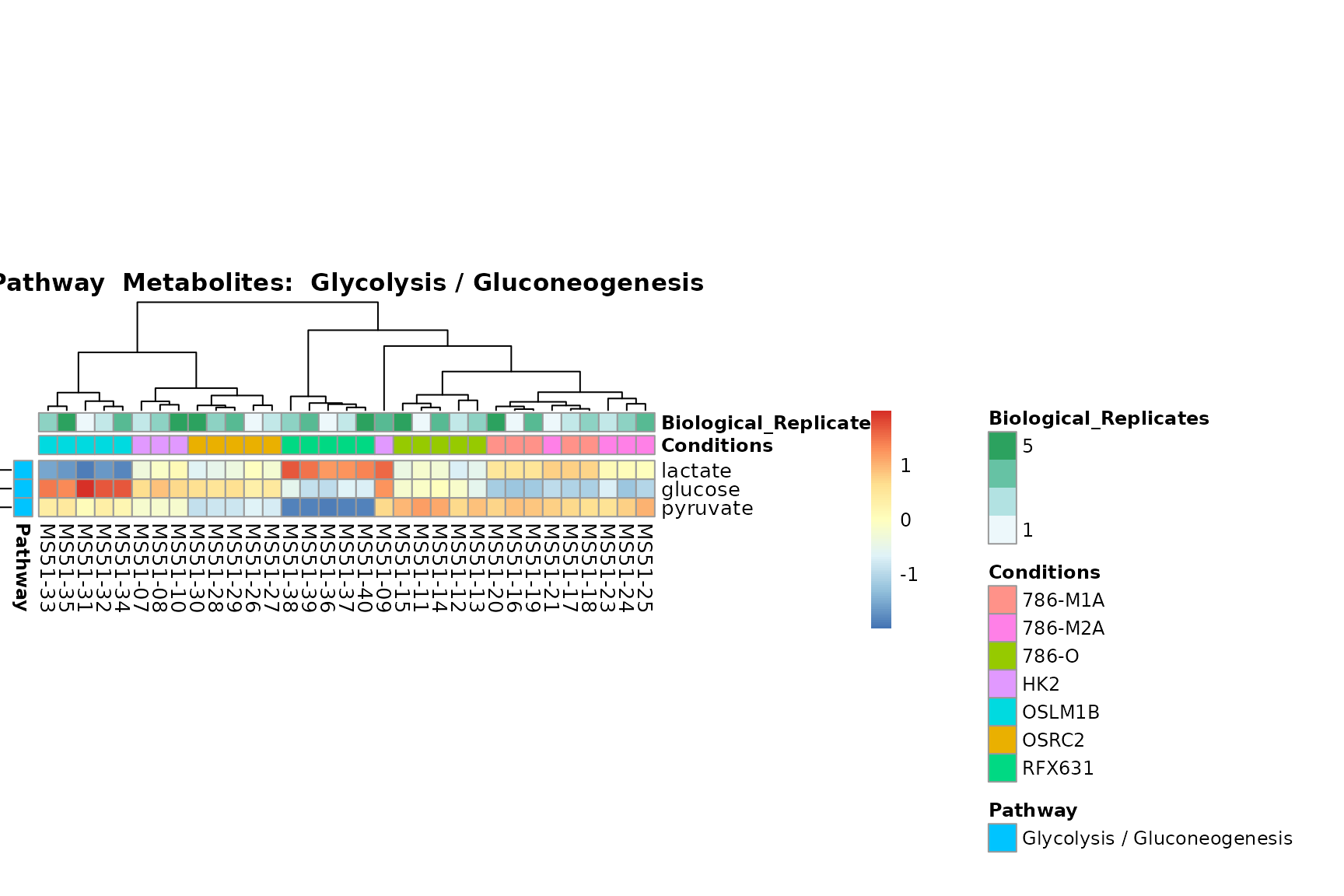
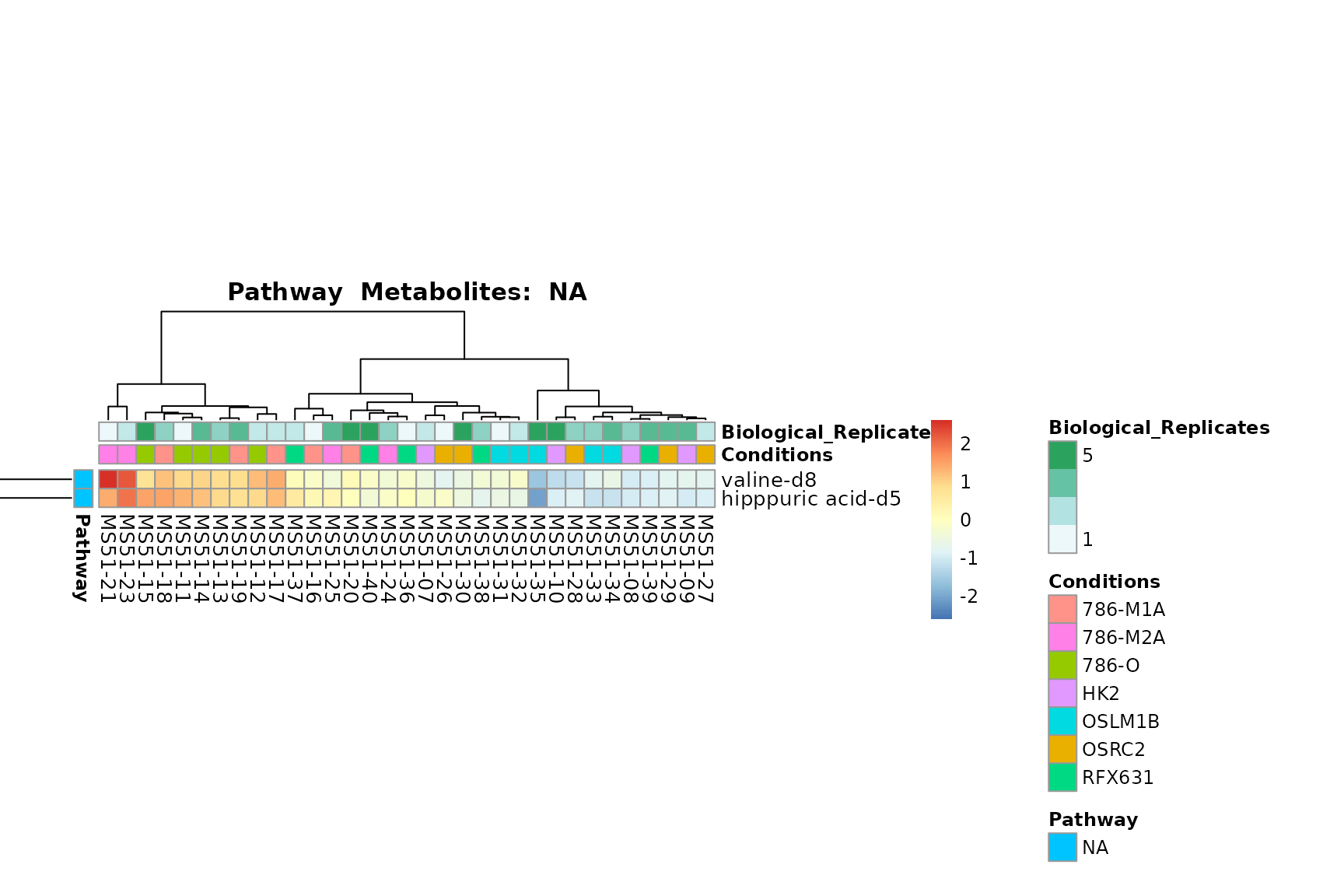

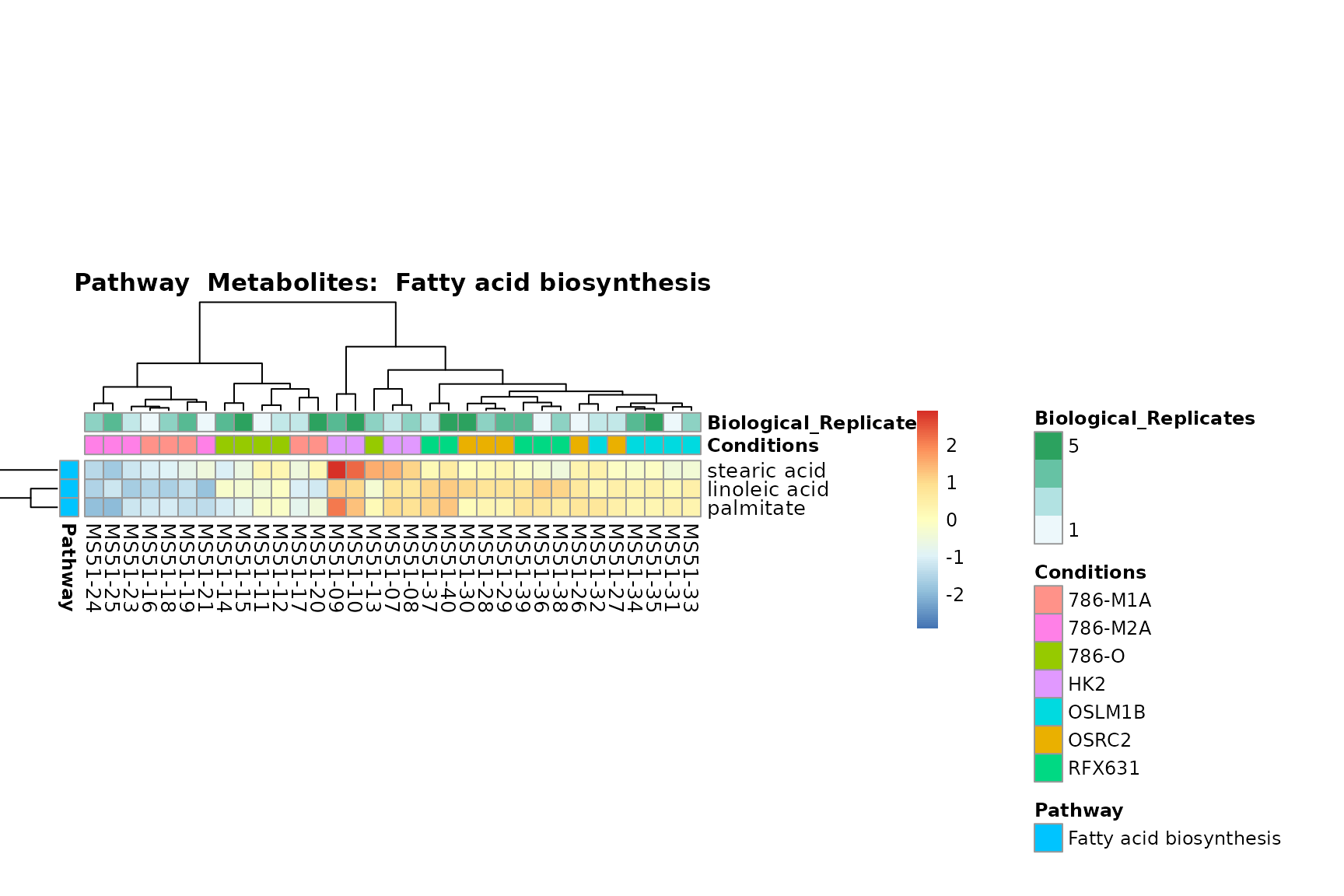
You can also choose to make individual plots for any Sample Metadata
using individual_Sample (e.g. in patients you may want to
plot male and female separately). Moreover, you can also use both at the
same time.
Superplots
Sometimes one might be interested to create individual plots for each
metabolite to understand the differences between specific conditions.
For this common plot types are bargraphs, boxplots or violin plots. As
input, we need a DF that contains the samples as rownames and the
features (=metabolites) as column names:
Input_Superplot <- Media_Preprocessed[,-c(1:4)]#remove columns that include Metadata such as cell type,...| valine-d8 | hipppuric acid-d5 | 2-hydroxyglutarate | 2-ketoglutarate | 3-Dehydro-L-threonate | acetylcarnitine | acetylcholine | |
|---|---|---|---|---|---|---|---|
| MS51-07 | 5049281744 | 30218506574 | -868601575 | 1530912430 | 28673393701 | -67547592848 | -496576017 |
| MS51-08 | -1441406385 | -11561761745 | 1108346025 | 3684365990 | 39313101752 | -234677799645 | -124297109 |
| MS51-09 | 2563904070 | -10237061776 | -189633550 | -9097094735 | -67782920598 | 218513442831 | 526295913 |
| MS51-10 | -6171779428 | -8419683053 | -50110899 | 3881816314 | -203574855 | 83711949661 | 94577213 |
| MS51-11 | 27378617230 | 123649127347 | 4311754839 | 23286535748 | -16721754062 | -434945308541 | -183109349 |
| MS51-12 | 31998154622 | 99965859879 | 808357705 | 19381509727 | -16349278172 | -583665678018 | -998475065 |
| MS51-13 | 26220971337 | 99972874145 | -235593734 | 19536285942 | -28131696536 | -645802166950 | -822951440 |
| MS51-14 | 27882417489 | 115689376712 | 1115432087 | 17101698081 | -26309877624 | -275257639669 | -1272434260 |
| MS51-15 | 24806237271 | 132343157722 | -1179339178 | 17846663766 | -40351828895 | -321374884275 | -1318142384 |
| MS51-16 | 11094140749 | 57354194293 | 5445671697 | 28056643231 | 9905712215 | -356638276041 | -929117918 |
| MS51-17 | 34144526544 | 116670253290 | 3041583921 | 22341957663 | -1871784883 | -379917701289 | -1217951807 |
| MS51-18 | 30843226841 | 131915991104 | 2364979960 | 19876068811 | -12106631836 | -297445077871 | -907725948 |
| MS51-19 | 26032777849 | 93842230196 | 600387750 | 22338213828 | -15996801077 | -325213646459 | -1216114751 |
| MS51-20 | 15413411654 | 47939629182 | 4007911817 | 20555474456 | 13695406411 | -438100232873 | -1430510133 |
| MS51-21 | 53001553101 | 127450060294 | 4809729749 | 39166236259 | -38896232803 | -125539074625 | -943874817 |
| MS51-23 | 47001796040 | 161667174110 | 6707051392 | 38517108736 | -6979130432 | -673387467221 | -664880164 |
| MS51-24 | 8217942926 | 37130736106 | 5857701472 | 39276732227 | -10740427138 | -333936377093 | -1206364609 |
| MS51-25 | 7242347148 | 57539536462 | 4568079831 | 40583987397 | -18377163802 | -282195052525 | -1314339238 |
| MS51-26 | 1631567949 | 36681096085 | 1121378276 | -2694080144 | -60644185117 | -198965081888 | -843128099 |
| MS51-27 | 1209476862 | -8930444074 | 3405336890 | -2060735139 | -51783244894 | -209186167980 | -365874316 |
| MS51-28 | -4518925715 | -2435870916 | 7007395646 | -2525310574 | -30952324005 | -408114545906 | -609942826 |
| MS51-29 | 1484077336 | -2058725307 | 3515115140 | -5164411612 | -42307838881 | -287356849930 | -912778912 |
| MS51-30 | 4368619286 | 15502685767 | -1093856576 | -4703700433 | -60704125766 | -260908005179 | -383882500 |
| MS51-31 | 7907484382 | 12534261776 | -2452294215 | -8563910085 | -75125518110 | -289811675340 | -918369787 |
| MS51-32 | 10026207227 | 8766901671 | 1953315759 | -7520737233 | -55168263224 | -890597864901 | -314644061 |
| MS51-33 | 1320088121 | -19794580878 | -541809575 | -6750277825 | -52231898984 | -770337735939 | -182991027 |
| MS51-34 | 4019534162 | -21662185450 | 1428924068 | -7578531520 | -47605477000 | -741976996697 | -550044215 |
| MS51-35 | -11674356179 | -75718957578 | 972005613 | -6094793780 | -57165852971 | -822161480737 | -503488121 |
| MS51-36 | 9892941550 | 46138316334 | 4954019648 | 5560697649 | -23867649475 | -71475573825 | -1119704368 |
| MS51-37 | 14414593887 | 78736973172 | -257560661 | -2341634721 | -51914744178 | -35405004462 | -687055540 |
| MS51-38 | 7716702900 | 3492507671 | -2307137058 | -4331637640 | -67900220172 | 230840109765 | -408157136 |
| MS51-39 | -583337199 | -9483365742 | 706088109 | -2757559248 | -56366820622 | -263196399957 | -227087862 |
| MS51-40 | 11097184979 | 24818171463 | 3186690665 | -4462779576 | -50563367720 | 145901488878 | -319389629 |
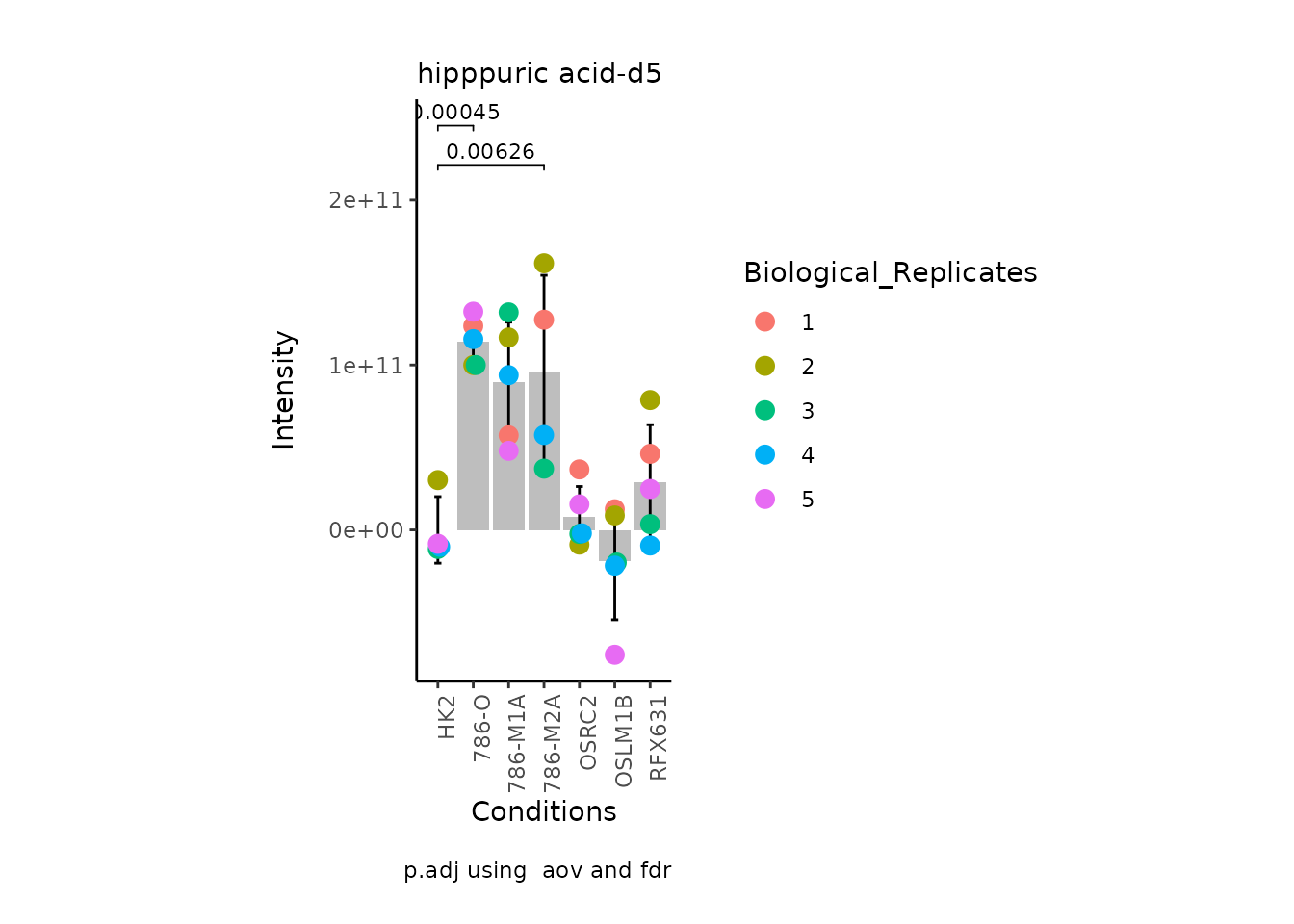
We also need the Metadata as we will need to know which conditions to
plot for together. If you have further information such as replicates or
patient ID, we can use this for the colour of the plotted samples per
condition as in the superplots style as described in by Lord et al (Lord et al. 2020).
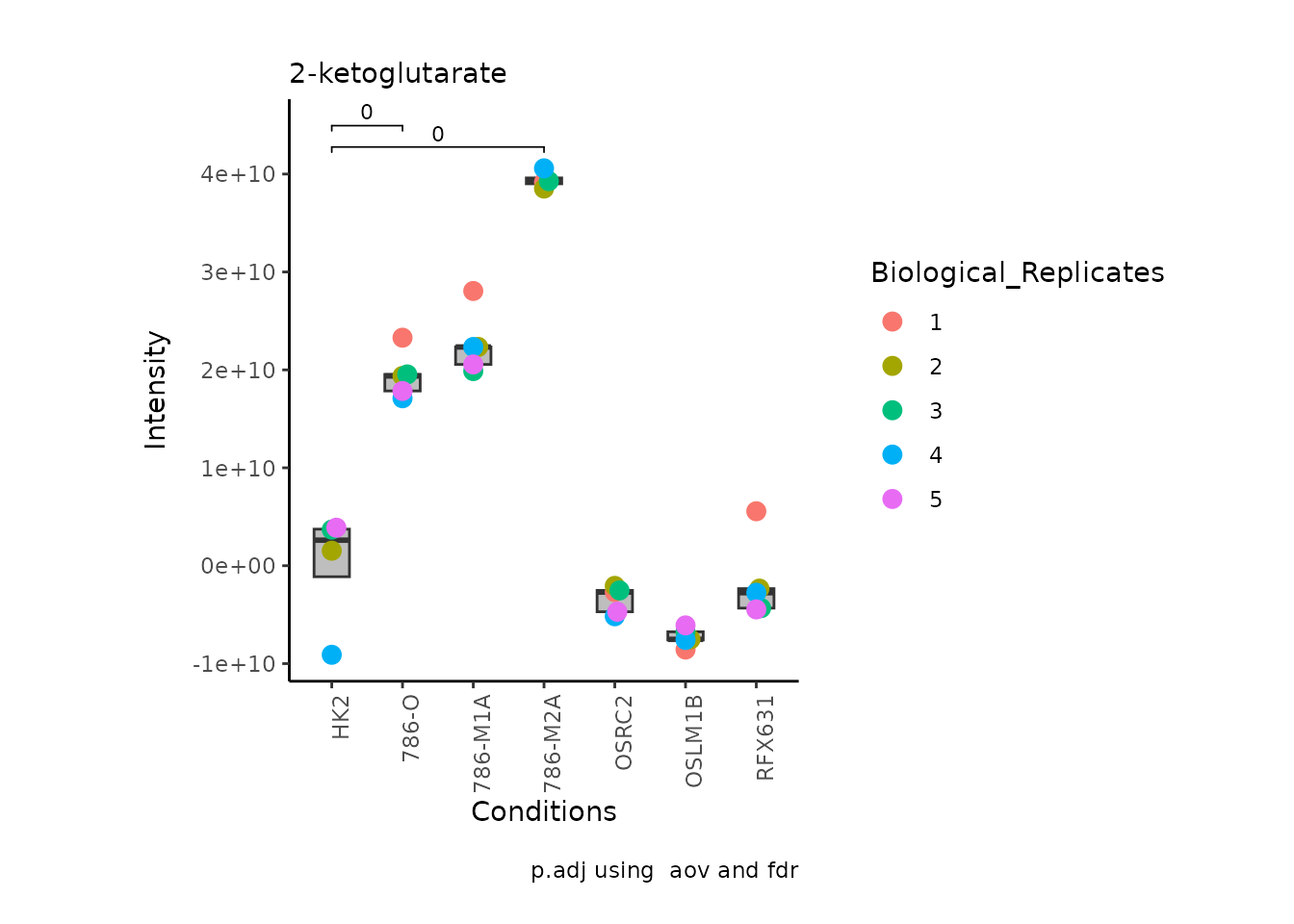
MetaProViz:::VizSuperplot(InputData =Input_Superplot[,c(1:6)],#We just plot six metabolites
SettingsFile_Sample =MetaData_Sample,
SettingsInfo = c(Conditions="Conditions", Superplot = "Biological_Replicates"),
PlotType = "Bar", #Bar, Box, Violin
PlotConditions = c("HK2", "786-O", "786-M1A", "786-M2A", "OSRC2", "OSLM1B", "RFX631"),#sets the order in which the samples should be plotted
StatComparisons = list(c(1,2),c(1,4)))#Stat comparisons to be included on the plot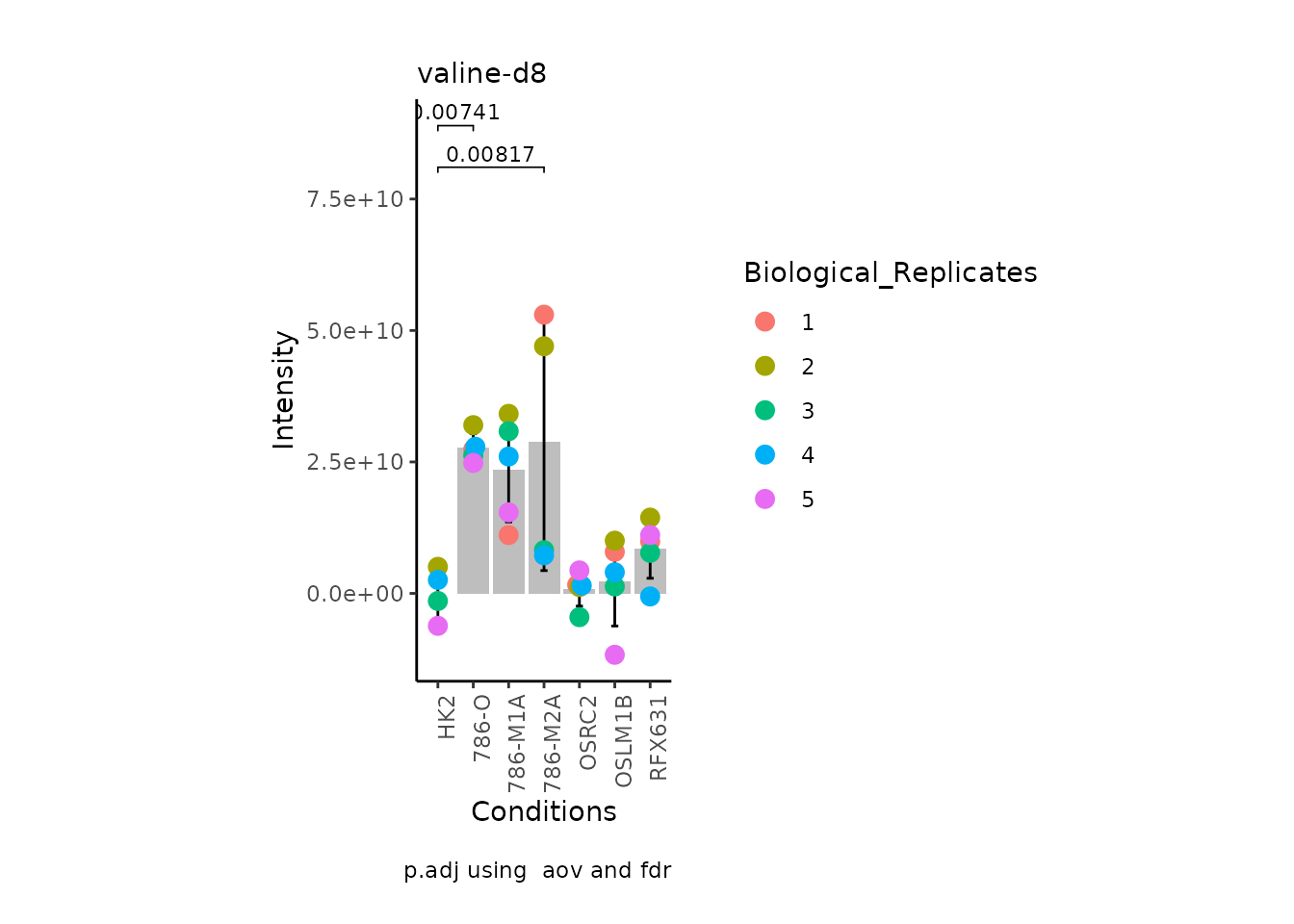
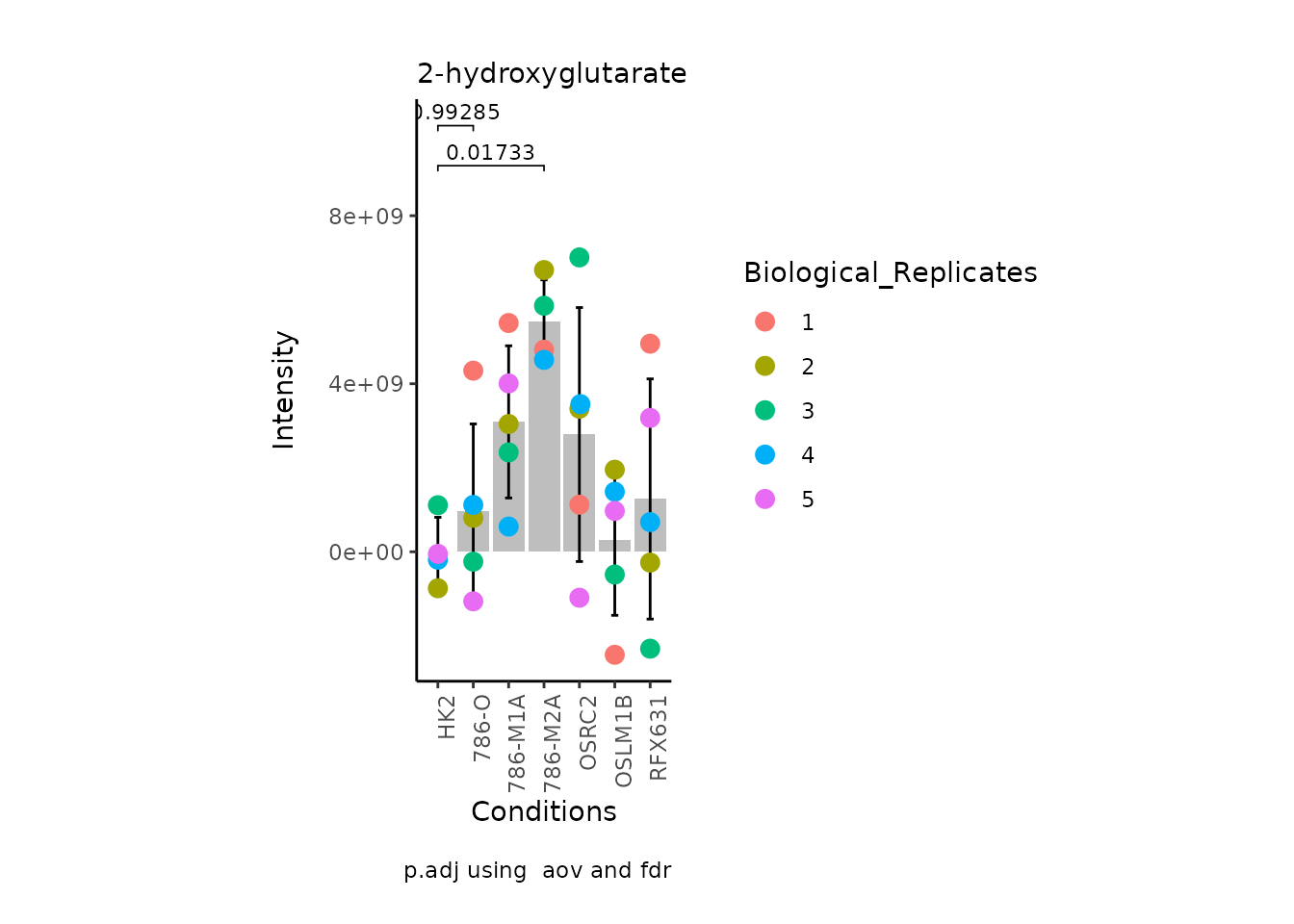
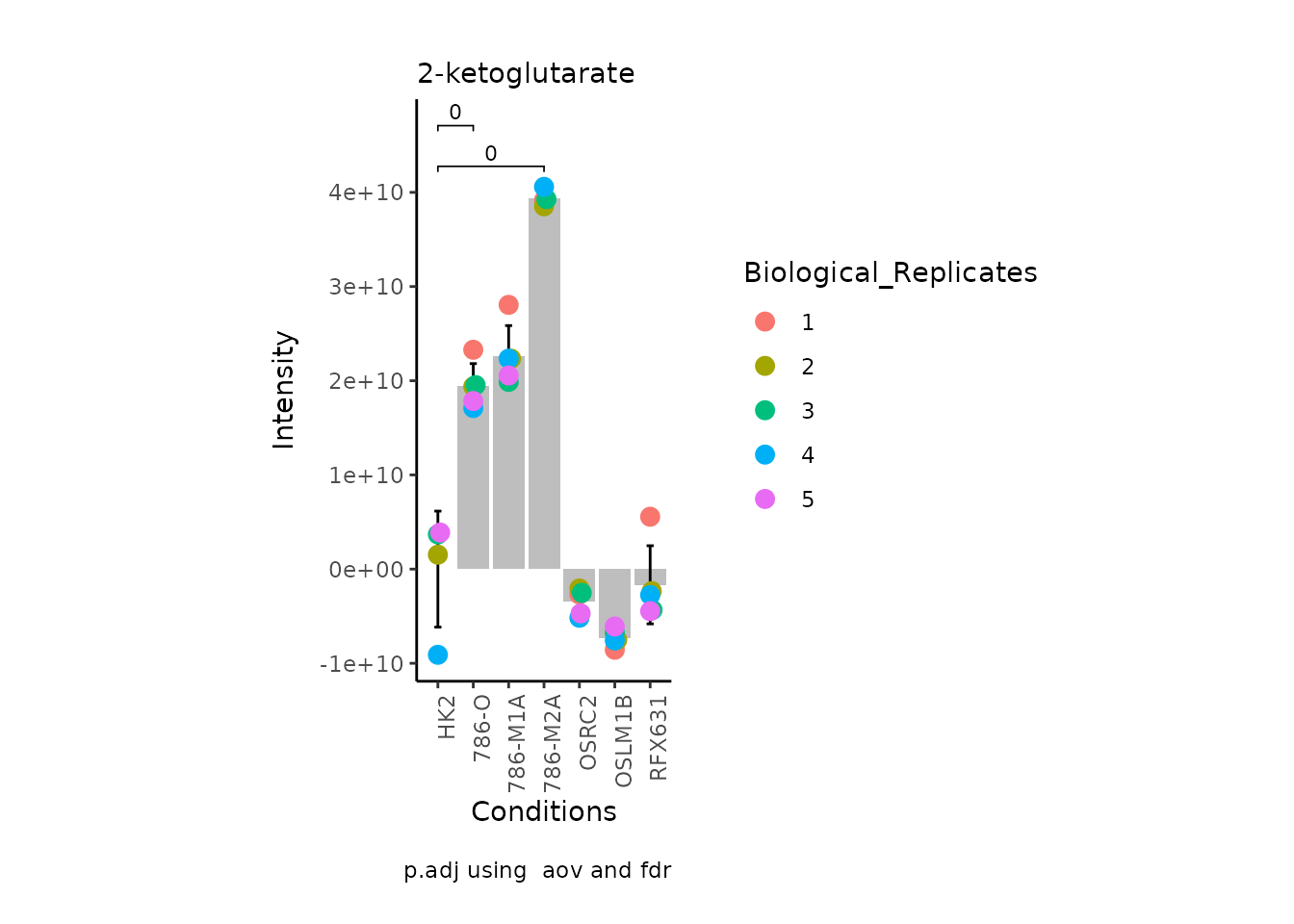
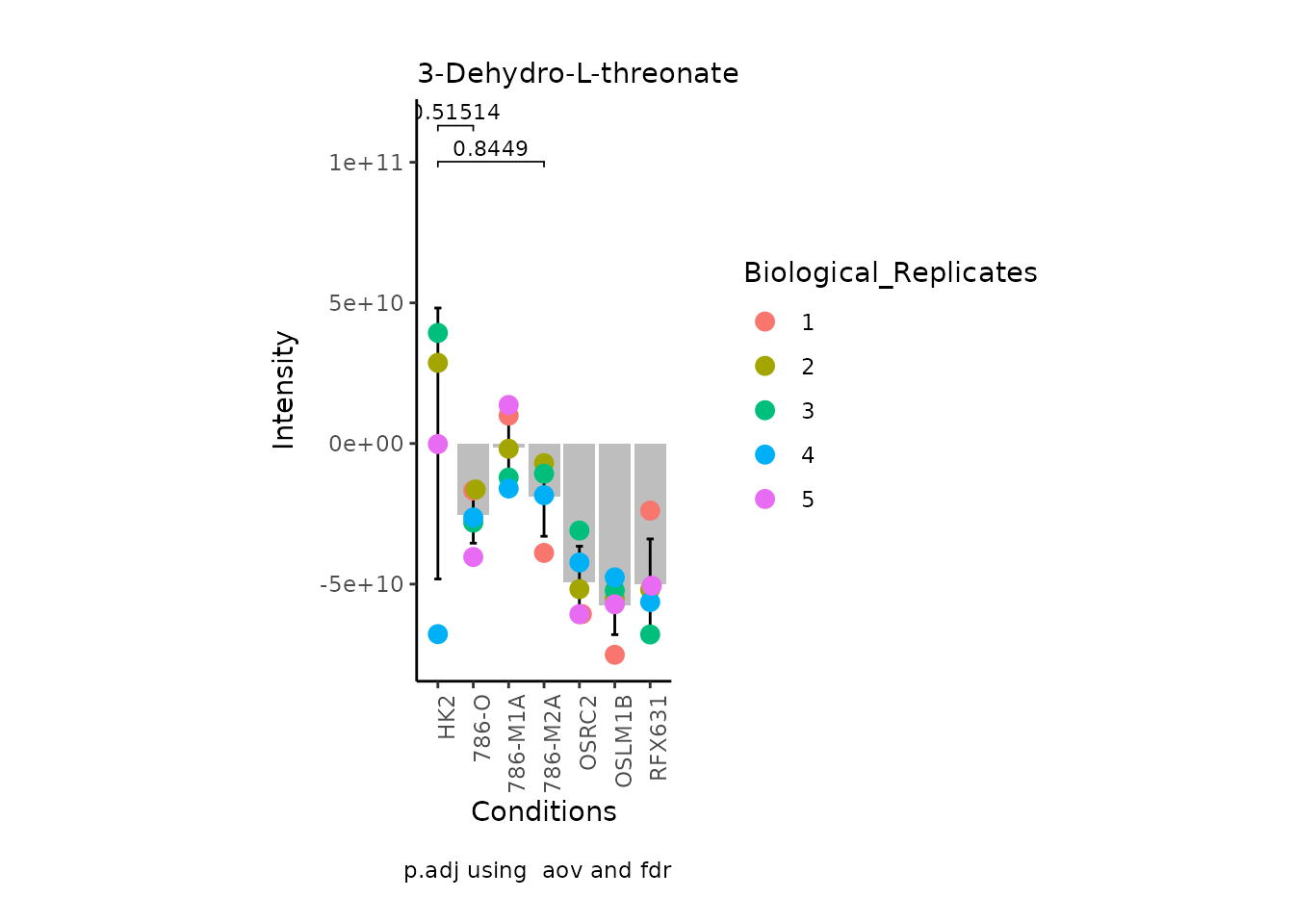
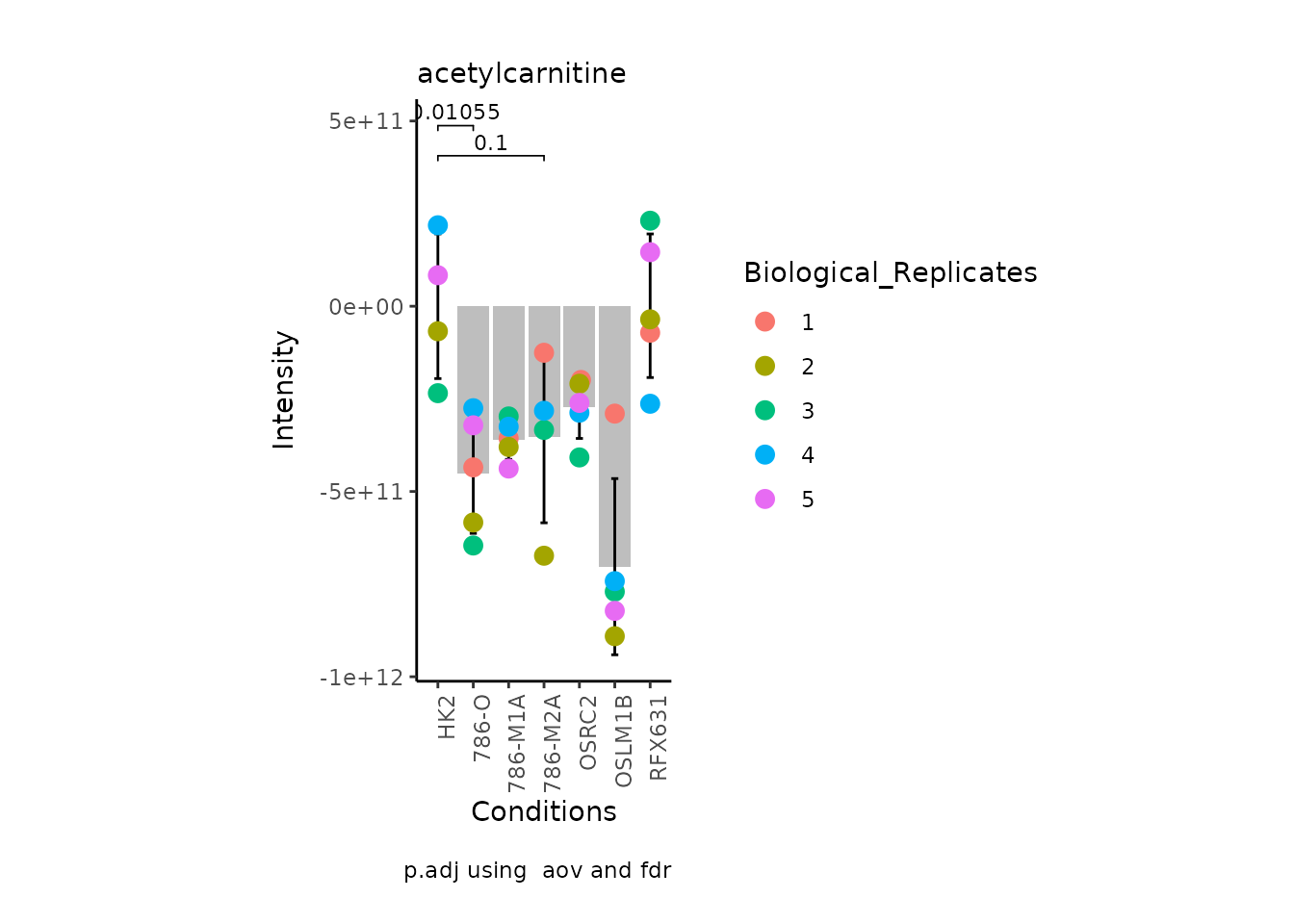
Now, if we for instance prefer boxplots over bargraphs we can simply
change the parameter PlotType:
MetaProViz:::VizSuperplot(InputData =Input_Superplot[,c(1:6)],#We just plot six metabolites
SettingsFile_Sample =MetaData_Sample,
SettingsInfo = c(Conditions="Conditions", Superplot = "Biological_Replicates"),
PlotType = "Box", #Bar, Box, Violin
PlotConditions = c("HK2", "786-O", "786-M1A", "786-M2A", "OSRC2", "OSLM1B", "RFX631"),#sets the order in which the samples should be plotted
StatComparisons = list(c(1,2),c(1,4)))#Stat comparisons to be included on the plot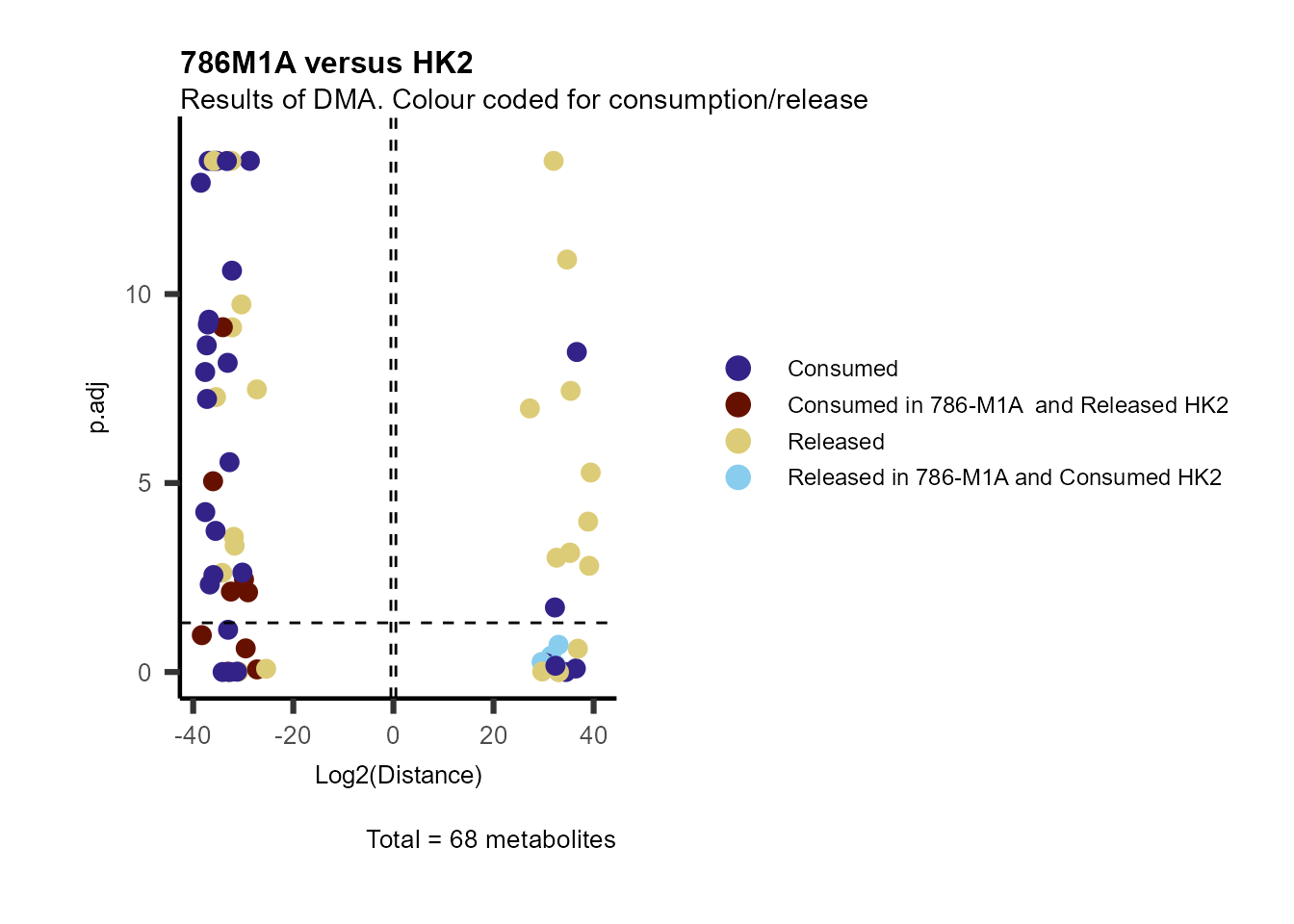
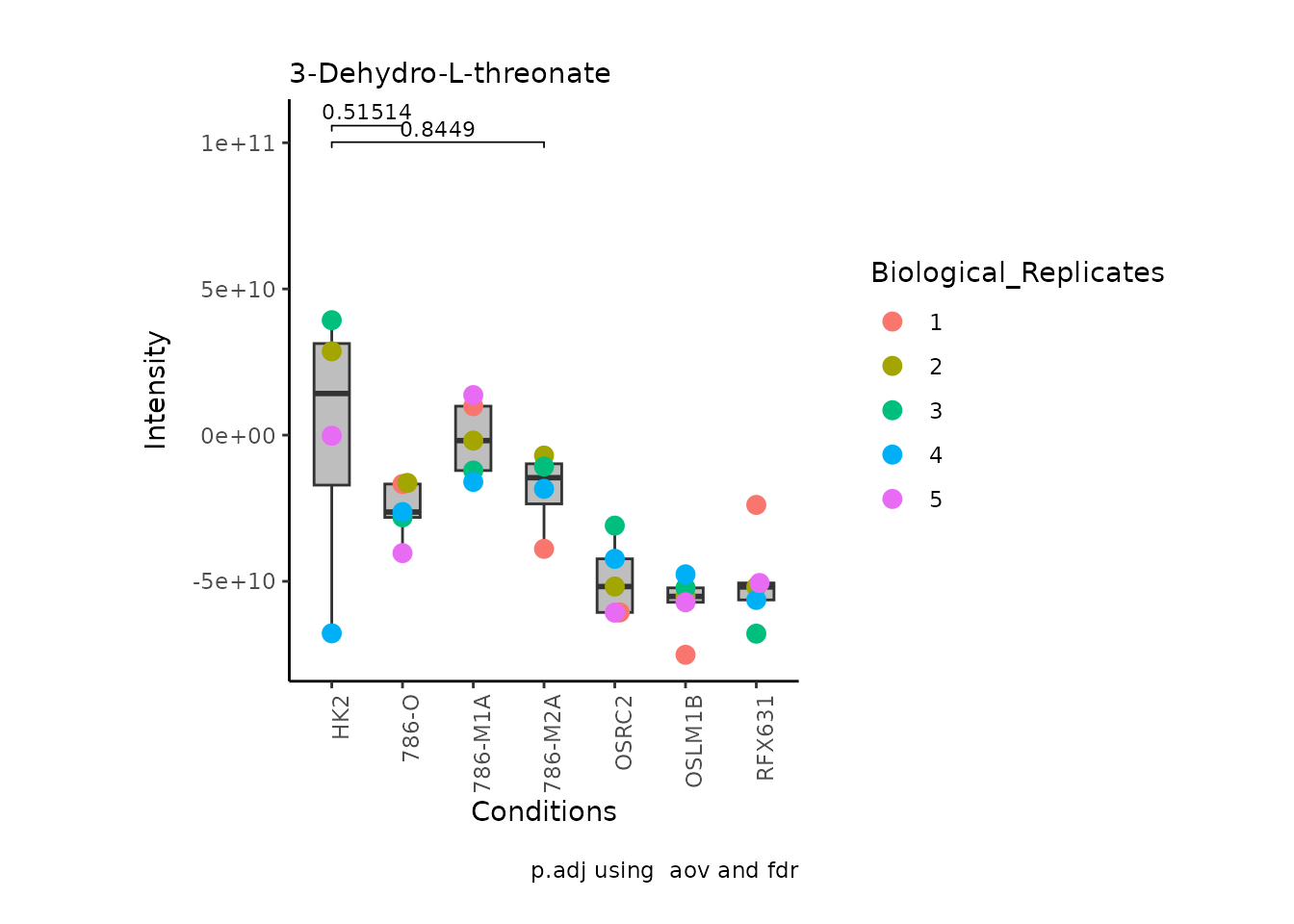
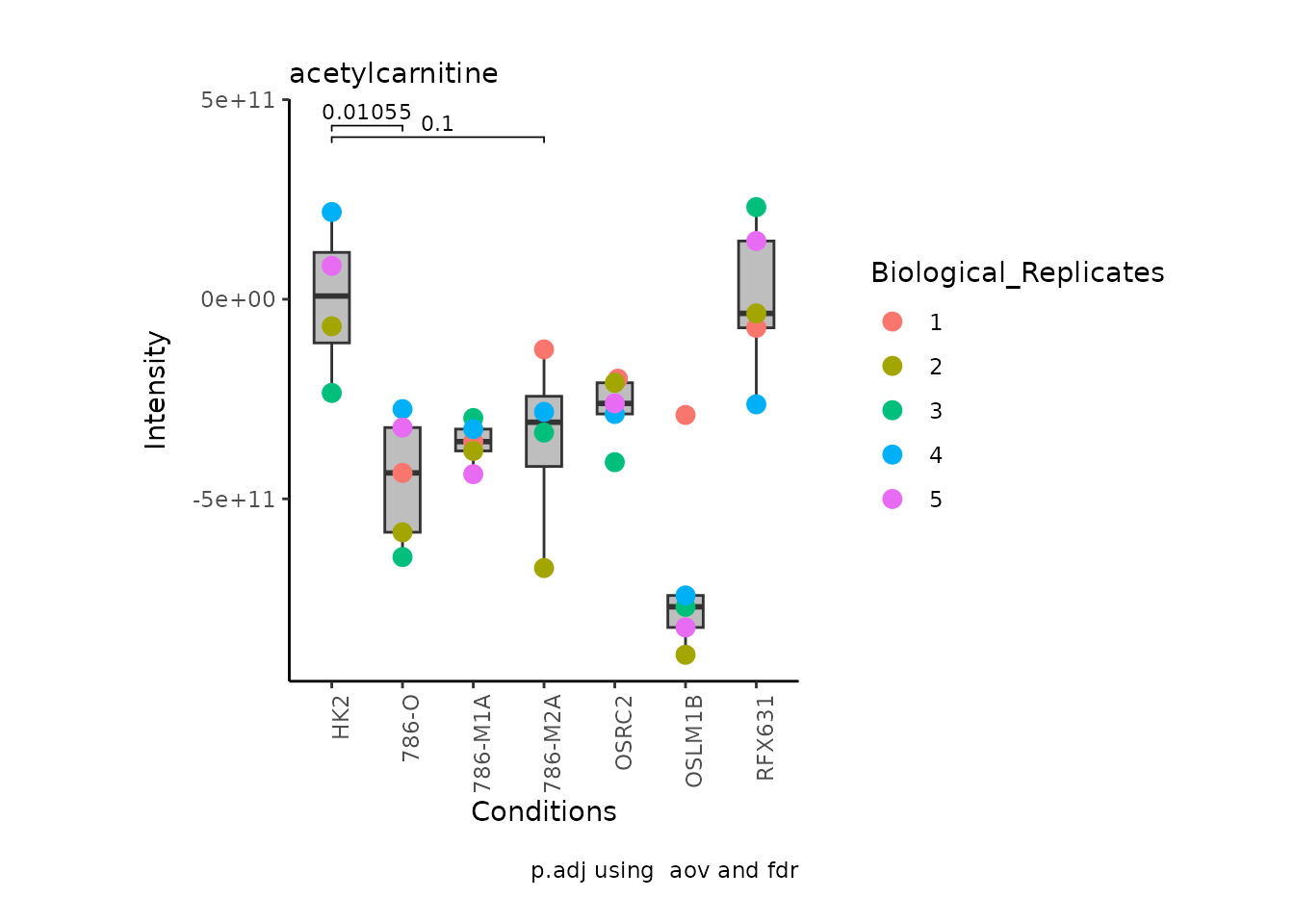
We can also change it to violin plots:
MetaProViz:::VizSuperplot(InputData =Input_Superplot[,c(1:6)],#We just plot six metabolites
SettingsFile_Sample =MetaData_Sample,
SettingsInfo = c(Conditions="Conditions", Superplot = "Biological_Replicates"),
PlotType = "Violin", #Bar, Box, Violin
PlotConditions = c("HK2", "786-O", "786-M1A", "786-M2A", "OSRC2", "OSLM1B", "RFX631"),#sets the order in which the samples should be plotted
StatComparisons = list(c(1,2),c(1,4)))#Stat comparisons to be included on the plot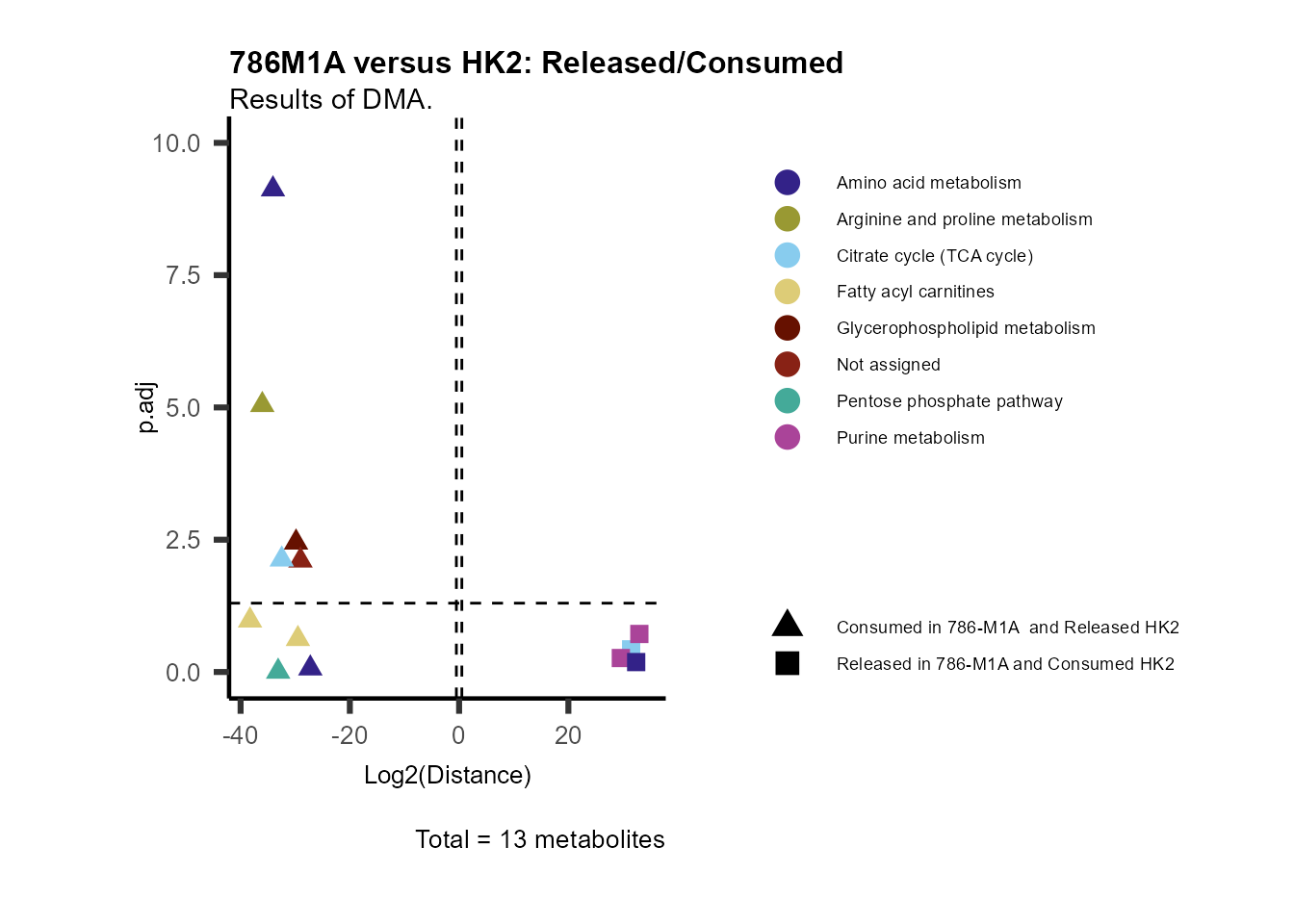

Volcano plot
In general,we have three differentPlot_Settings, which
will also be used for other plot types such as lollipop graphs.1. "Standard" is the standard version of the
plot, with one dataset being plotted.2. "Conditions" here two or more datasets will
be plotted together. How datasets can be plotted together depends on the
plot type.3. "PEA" stands for Pathway Enrichment
Analysis, and is used if the results of an GSE analysis should be
plotted as here the figure legends will be adapted. You can find an
example for this in the vignette
Standard MetabolomicsHere we will look at all the different options we have to display our results from the different analysis, which will help us to interpret our results as this can be sometimes difficult to do from the many data tables.
Just a quick reminder, how the input data look like:
1. Results of Differential Metabolite Analysis (DMA): Log2(Distance) and stats:
| Metabolite | Log2(Distance) | p.adj | t.val | Mean_786-M1A | CoRe_786-M1A |
|---|---|---|---|---|---|
| 2-hydroxyglutarate | 31.52594 | 0.3447870 | -3092107029 | 3092107028.86536 | Released |
| 2-ketoglutarate | 34.39775 | 0.0000000 | -22633671598 | 22633671597.7763 | Released |
| 3-Dehydro-L-threonate | -30.24765 | 0.9999999 | 1274819834 | -1274819834.18323 | Consumed |
| acetylcarnitine | -38.38705 | 0.0653736 | 359462986907 | -359462986906.515 | Consumed |
| acetylcholine | -30.08675 | 0.0004996 | 1140284111 | -1140284111.31361 | Consumed |
| acetylornithine | 32.65274 | 0.0012793 | -6752323351 | 6752323351.13854 | Released |
| aconitate | -32.58145 | 0.0000000 | 6426780519 | -6426780518.51619 | Consumed |
Standard
Here we will first look into the results from the differential
analysis (see section DMA above) for the comparison of
786-M1A_vs_HK2:
# Run with default parameter --> only need to provide Input_data and the title we like
MetaProViz::VizVolcano(InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)")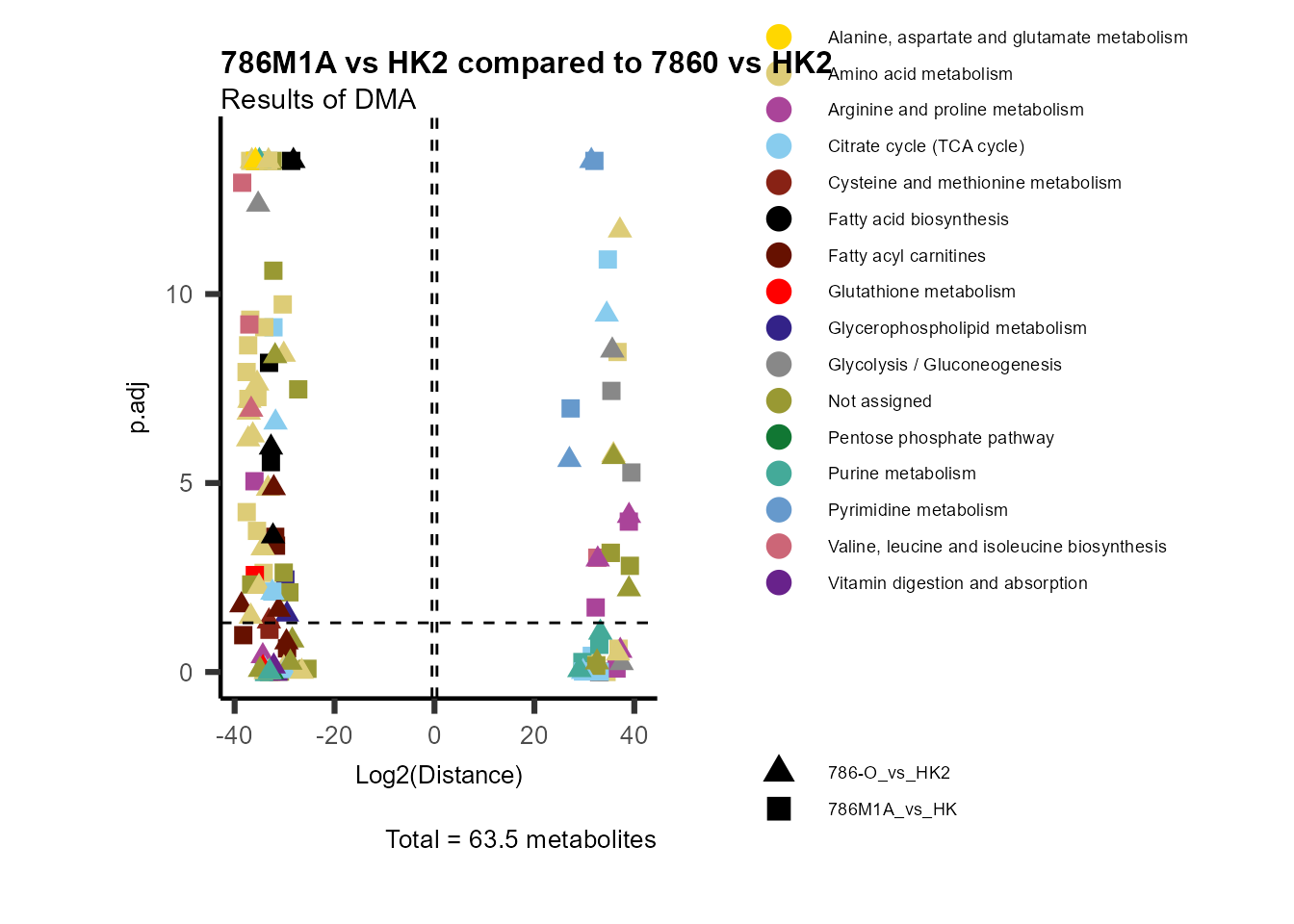
Figure: Standard figure displaying DMA results.
If you seek to plot the metabolite names you can change the paramter
SelectLab from its default (SelectLab="") to
NULL and the metabolite names will be plotted randomly.
# Run with default parameter --> only need to provide Input_data and the title we like
MetaProViz::VizVolcano(InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
SelectLab = NULL)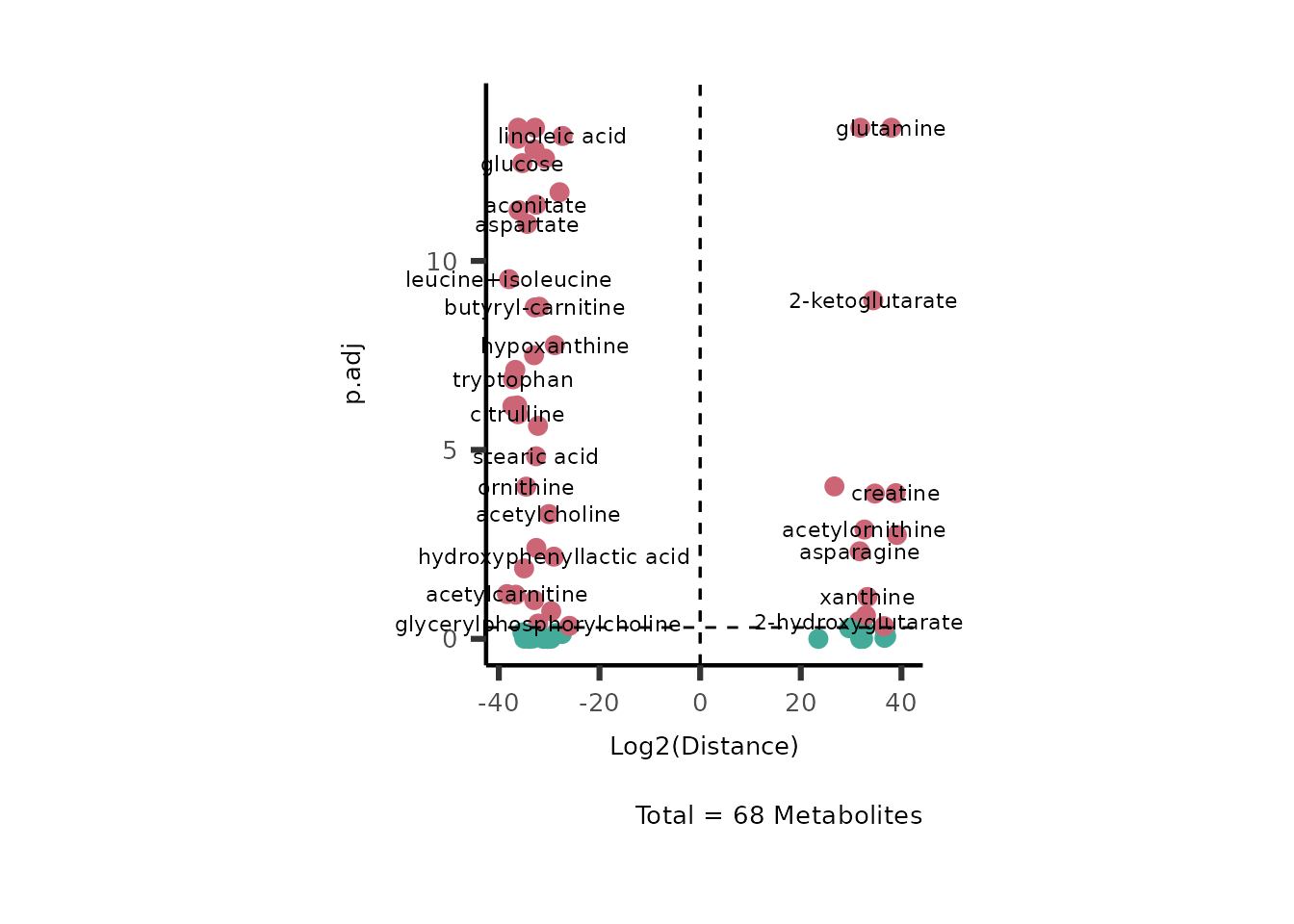
Figure: Standard figure displaying DMA results.
With the parameter SelectLab you can also pass a vector
with Metabolite names that should be labeled:
# Run with default parameter --> only need to provide Input_data and the title we like
MetaProViz::VizVolcano(InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
SelectLab = c("histidine", "phenylalanine", "lactate"))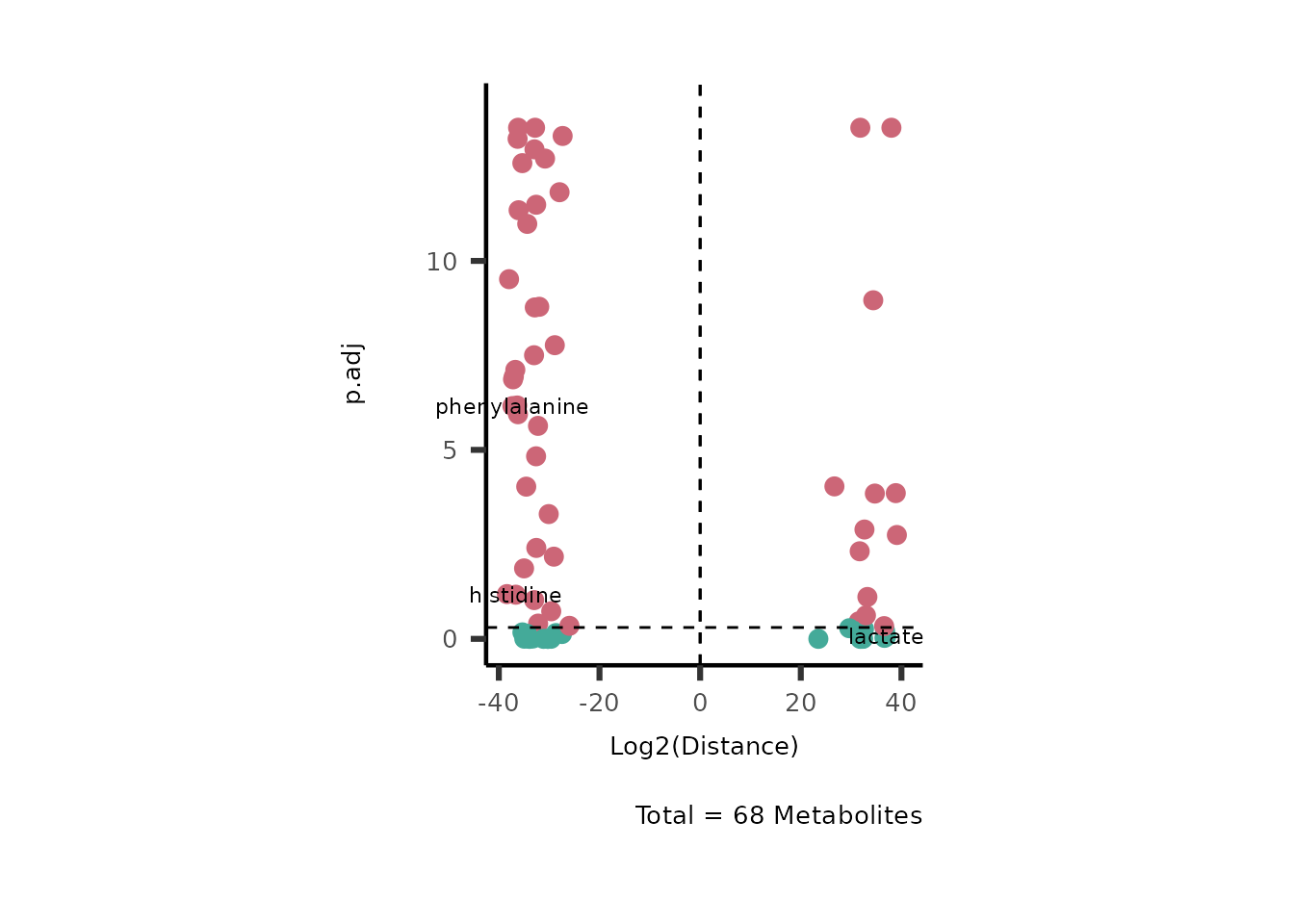
Figure: Standard figure displaying DMA results.
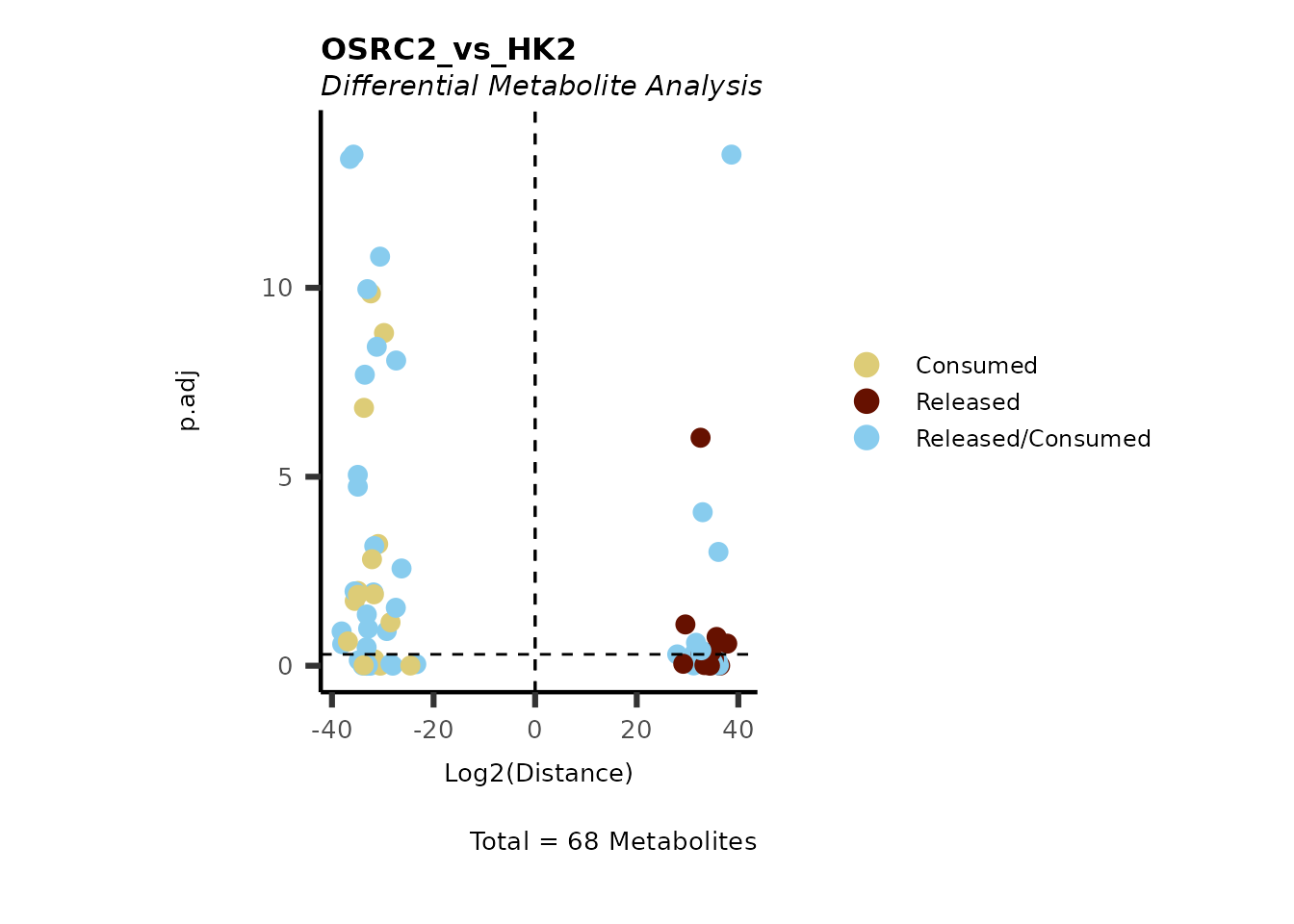
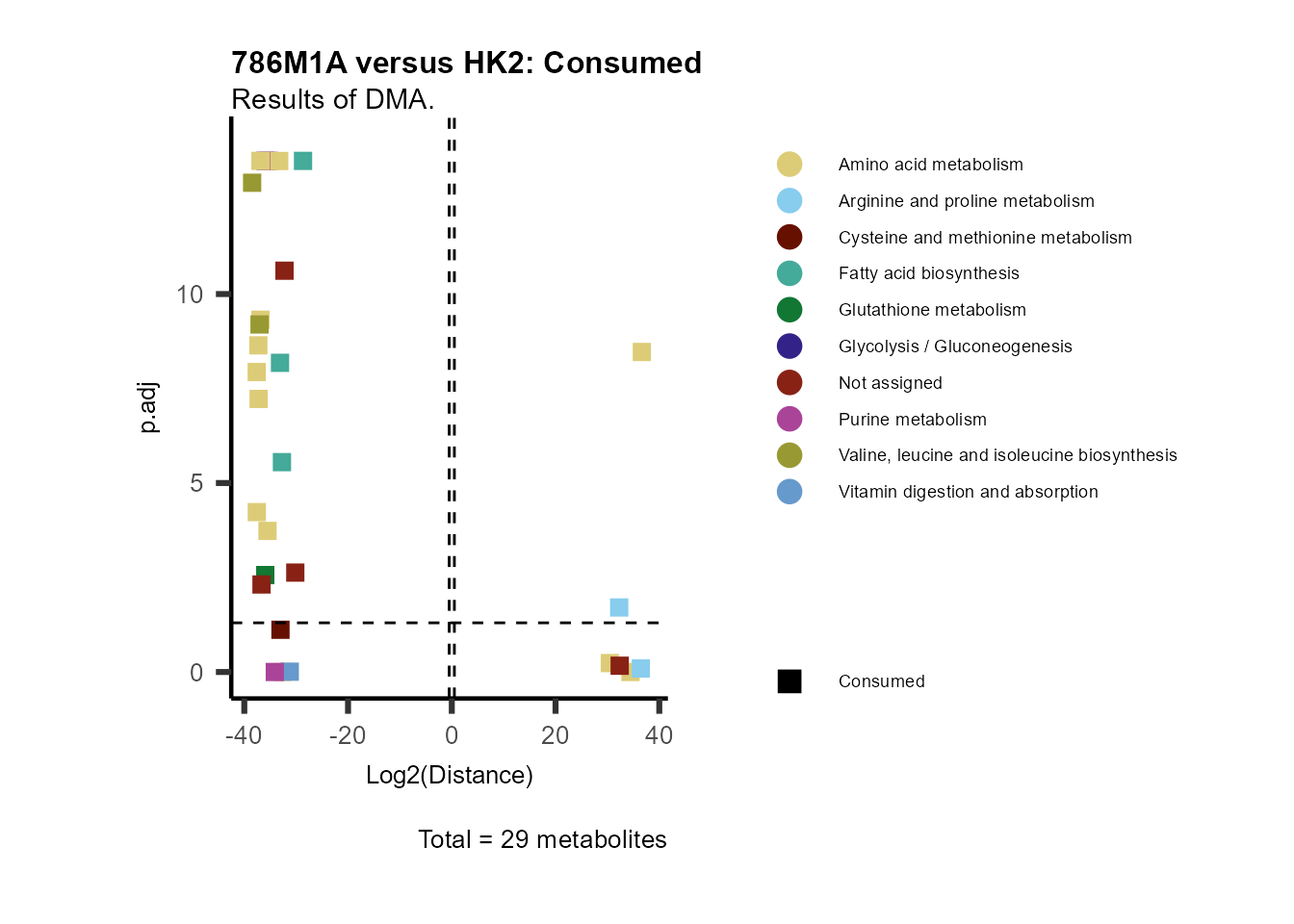
As explained above, when analyzing CoRe data it is important to take
into account if a metabolite is consumed or released. we can use this
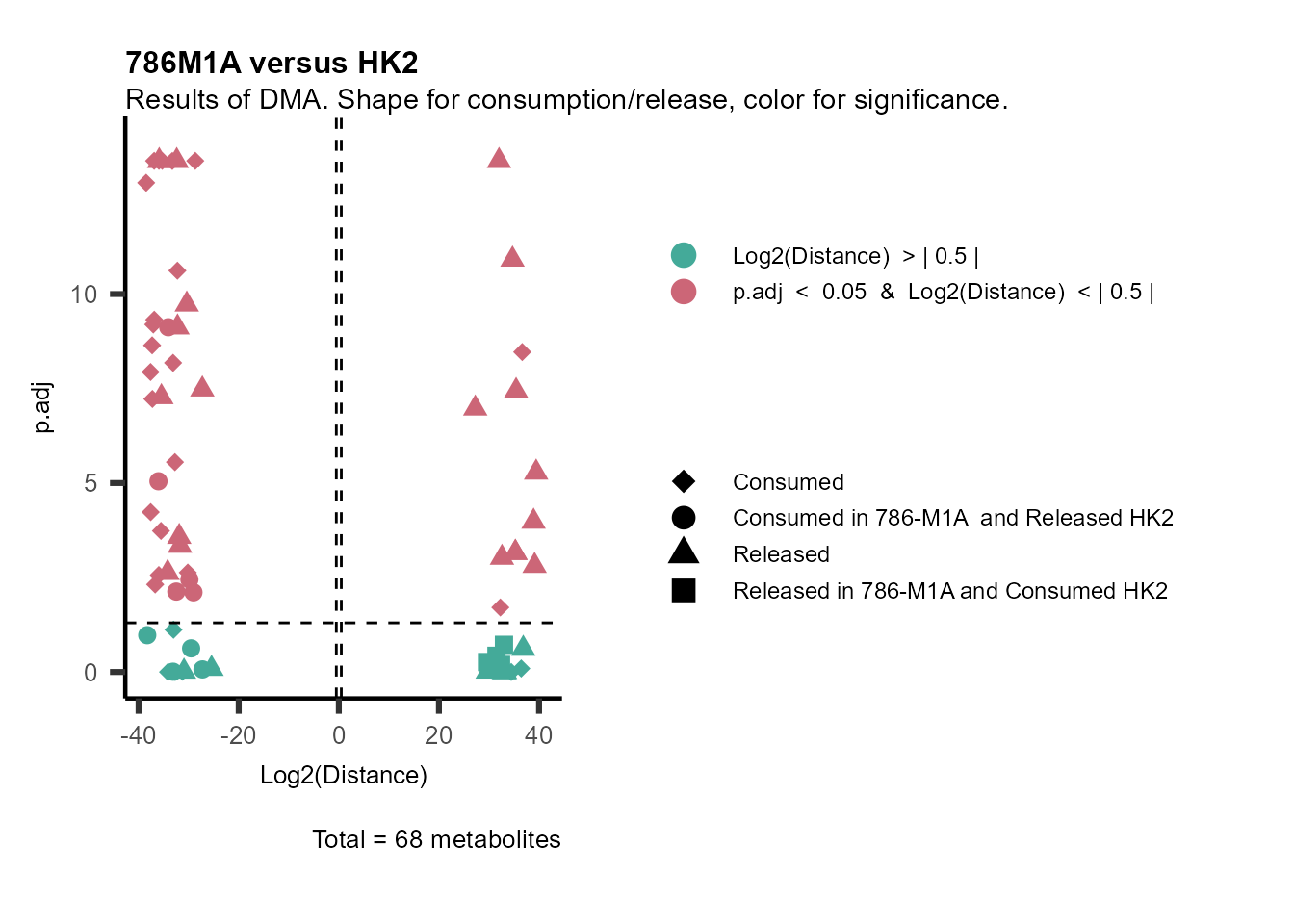
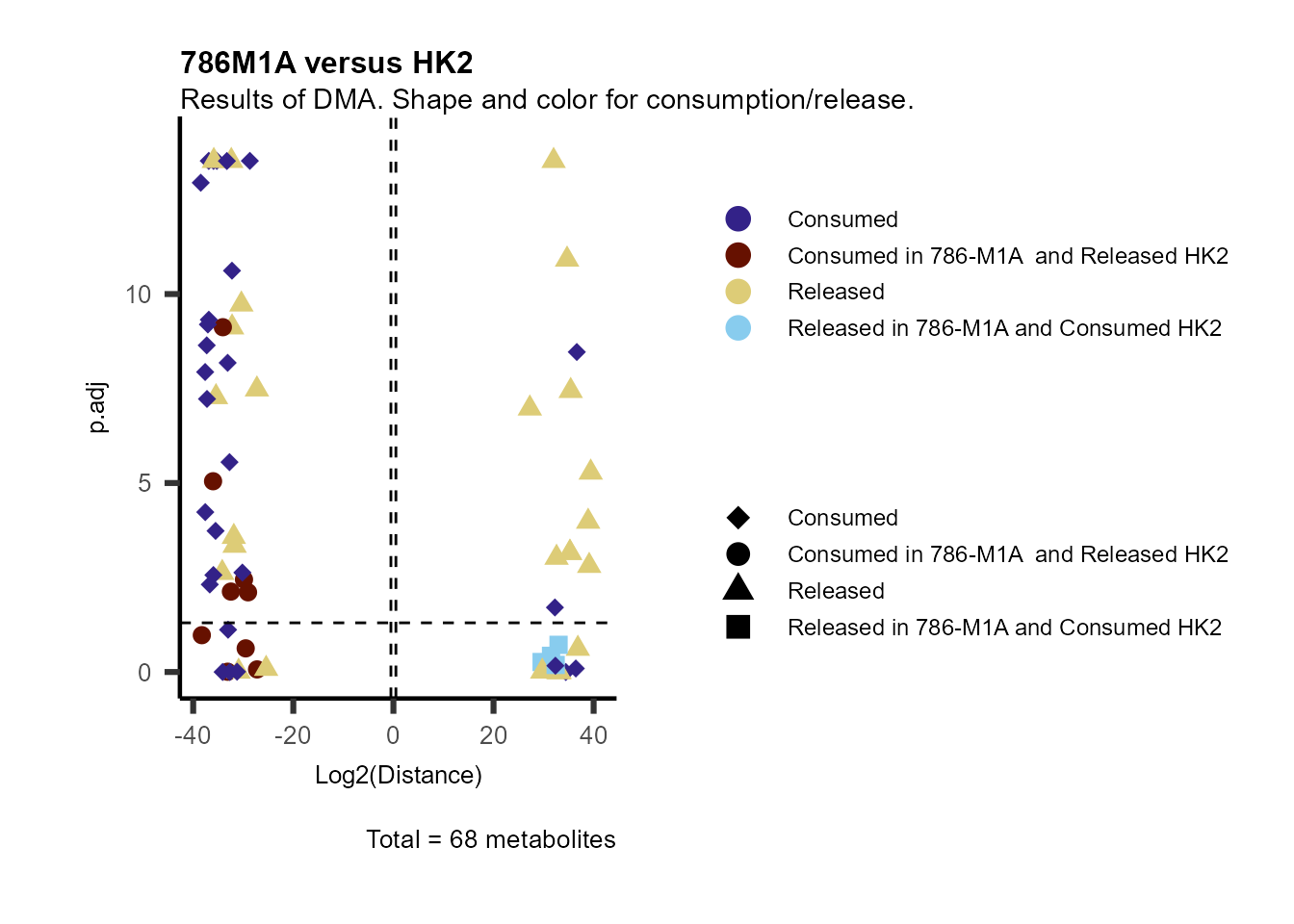
information to colour code and or shape the metabolites on the
plot.
For this we need to add this information into the Metadata_Metabolite
file:
# colour for consumption and release: For this we need to add this information into the Metadata_Metabolite file
MetaData_Metab <- merge(MappingInfo%>%rownames_to_column("Metabolite"), DMA_786M1A_vs_HK2[,c(1,6,8:10)], by="Metabolite", all.y=TRUE)%>%
column_to_rownames("Metabolite")| HMDB | KEGG.ID | KEGGCompound | Pathway | CoRe_786-M1A | CoRe_HK2 | CoRe_specific | CoRe | |
|---|---|---|---|---|---|---|---|---|
| 3-Dehydro-L-threonate | NA | NA | NA | Amino acid metabolism | Consumed | Consumed | Consumed | Consumed |
| acetylcarnitine | HMDB0000201 | C02571 | O-Acetylcarnitine | Fatty acyl carnitines | Consumed | Released | Consumed in 786-M1A and Released HK2 | Released/Consumed |
| acetylornithine | HMDB0003357 | C00437 | N-Acetylornithine | Arginine and proline metabolism | Released | Consumed | Released in 786-M1A and Consumed HK2 | Released/Consumed |
| glutamate | HMDB0000148 | C00025 | L-Glutamate | Amino acid metabolism | Released | Consumed | Released in 786-M1A and Consumed HK2 | Released/Consumed |
| glutamine | HMDB0000641 | C00064 | L-Glutamine | Amino acid metabolism | Released | Consumed | Released in 786-M1A and Consumed HK2 | Released/Consumed |
| glycerylphosphorylcholine | HMDB0252858 | NA | NA | Not assigned | Consumed | Consumed | Consumed | Consumed |
Now we can make the different plots:
#Now we need to add our Plot_SettingsFile and the Plot_SettingsInfo:
MetaProViz::VizVolcano(PlotSettings="Standard",
SettingsInfo= c(color="CoRe_specific"),
SettingsFile_Metab= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
PlotName= "786M1A versus HK2",
Subtitle= "Results of DMA. Colour coded for consumption/release" )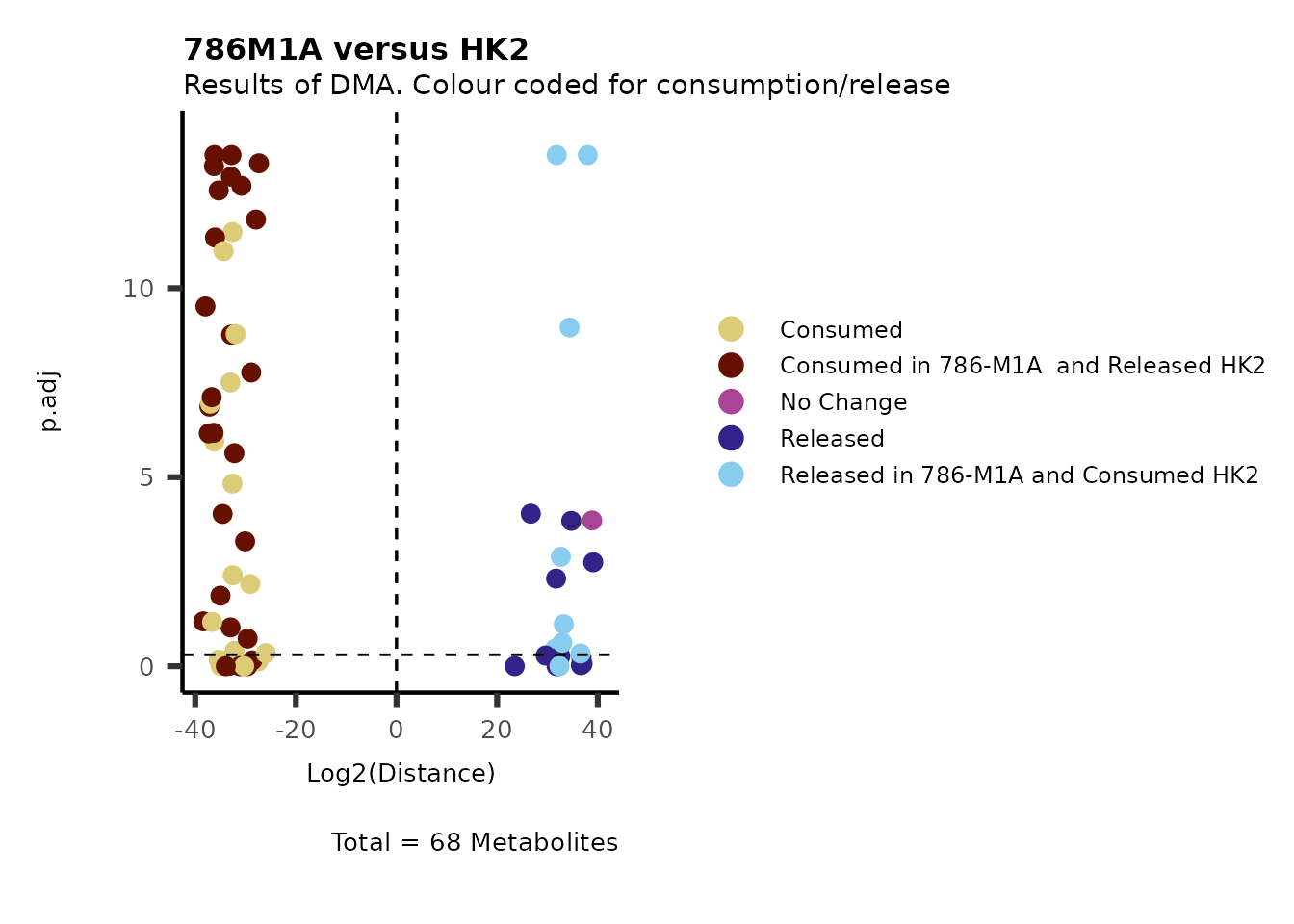
Figure: Standard figure displaying DMA results.
#If we want to use the shape instead of the colour for the cluster info, we can just change our Plot_SettingsInfo
MetaProViz::VizVolcano(PlotSettings="Standard",
SettingsInfo= c(shape="CoRe_specific"),
SettingsFile= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
PlotName= "786M1A versus HK2",
Subtitle= "Results of DMA. Shape for consumption/release, color for significance." )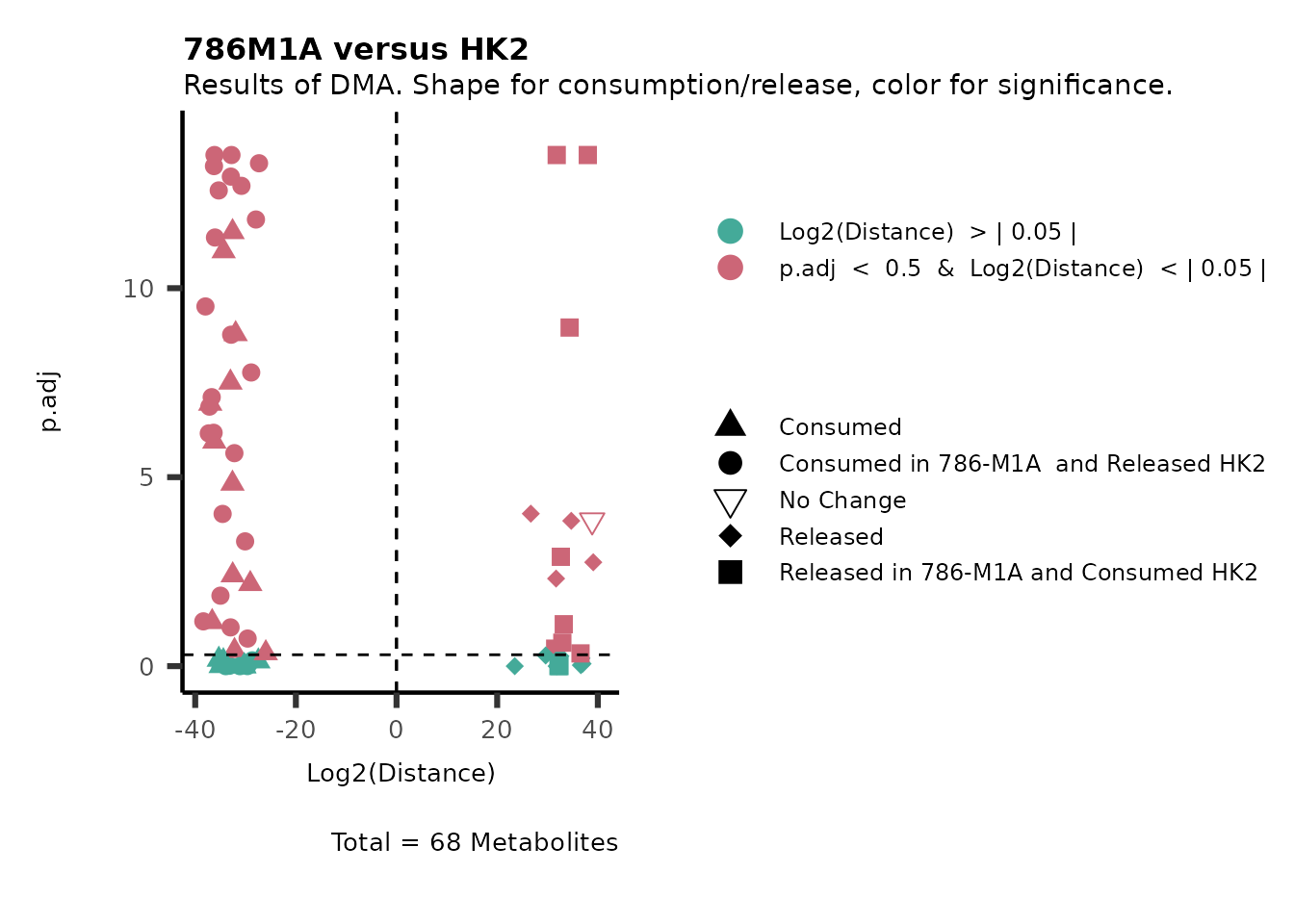
Figure: Standard figure displaying DMA results.
#Of course, we can also adapt both, color and shape for the same parameter:
MetaProViz::VizVolcano(PlotSettings="Standard",
SettingsInfo= c(shape="CoRe_specific", color="CoRe_specific"),
SettingsFile= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
PlotName= "786M1A versus HK2",
Subtitle= "Results of DMA. Shape and color for consumption/release." )
Figure: Standard figure displaying DMA results.
Of course, here we may also want an individual plot for each of the
consumption/release metabolites.
#individual plot for each metabolite behaviour:
MetaProViz::VizVolcano(PlotSettings="Standard",
SettingsInfo= c(individual="CoRe", shape="CoRe_specific"),
SettingsFile= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
PlotName= "786M1A versus HK2",
Subtitle= "Results of DMA." )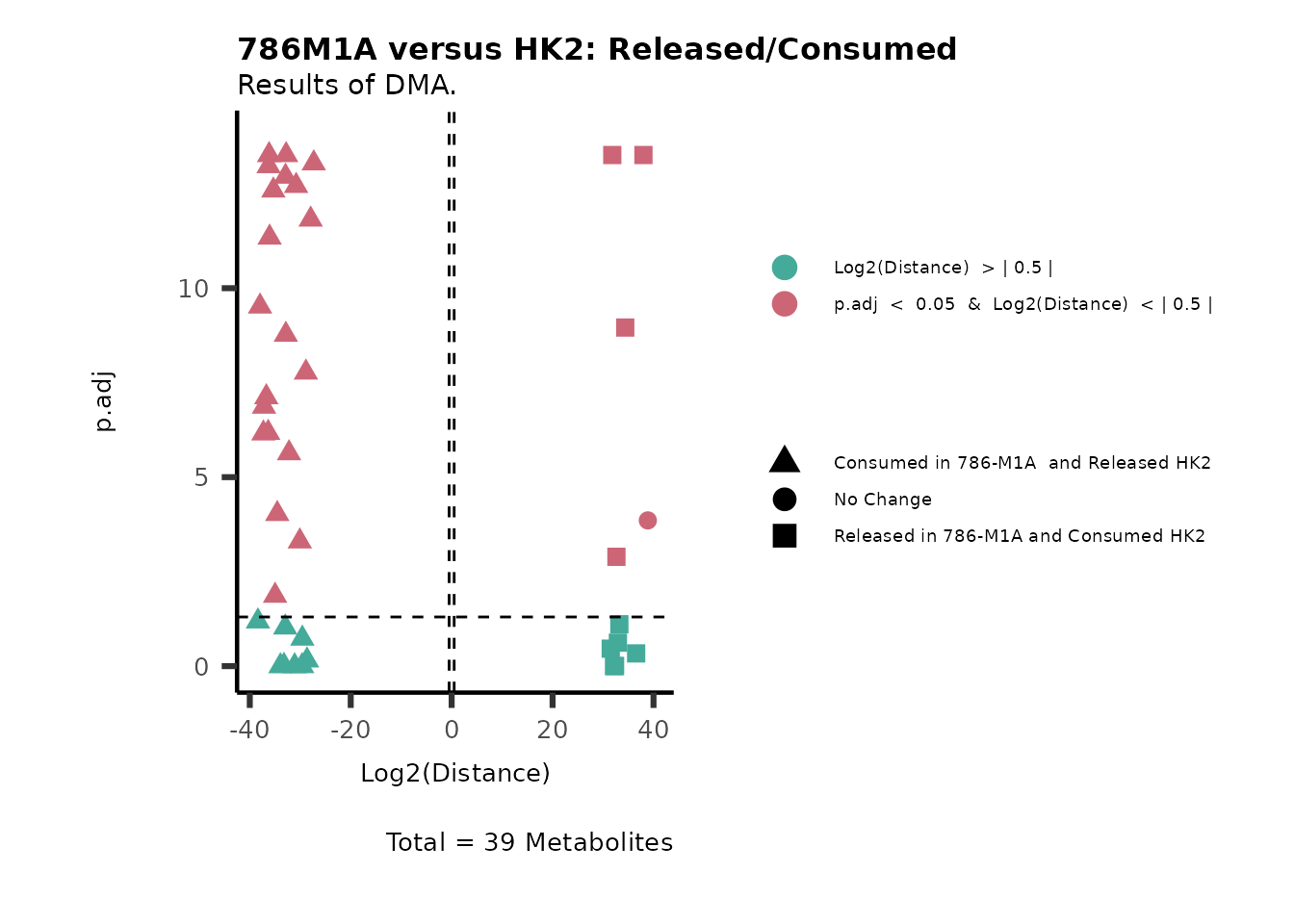
Figure: Standard figure displaying DMA results.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).
#> Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).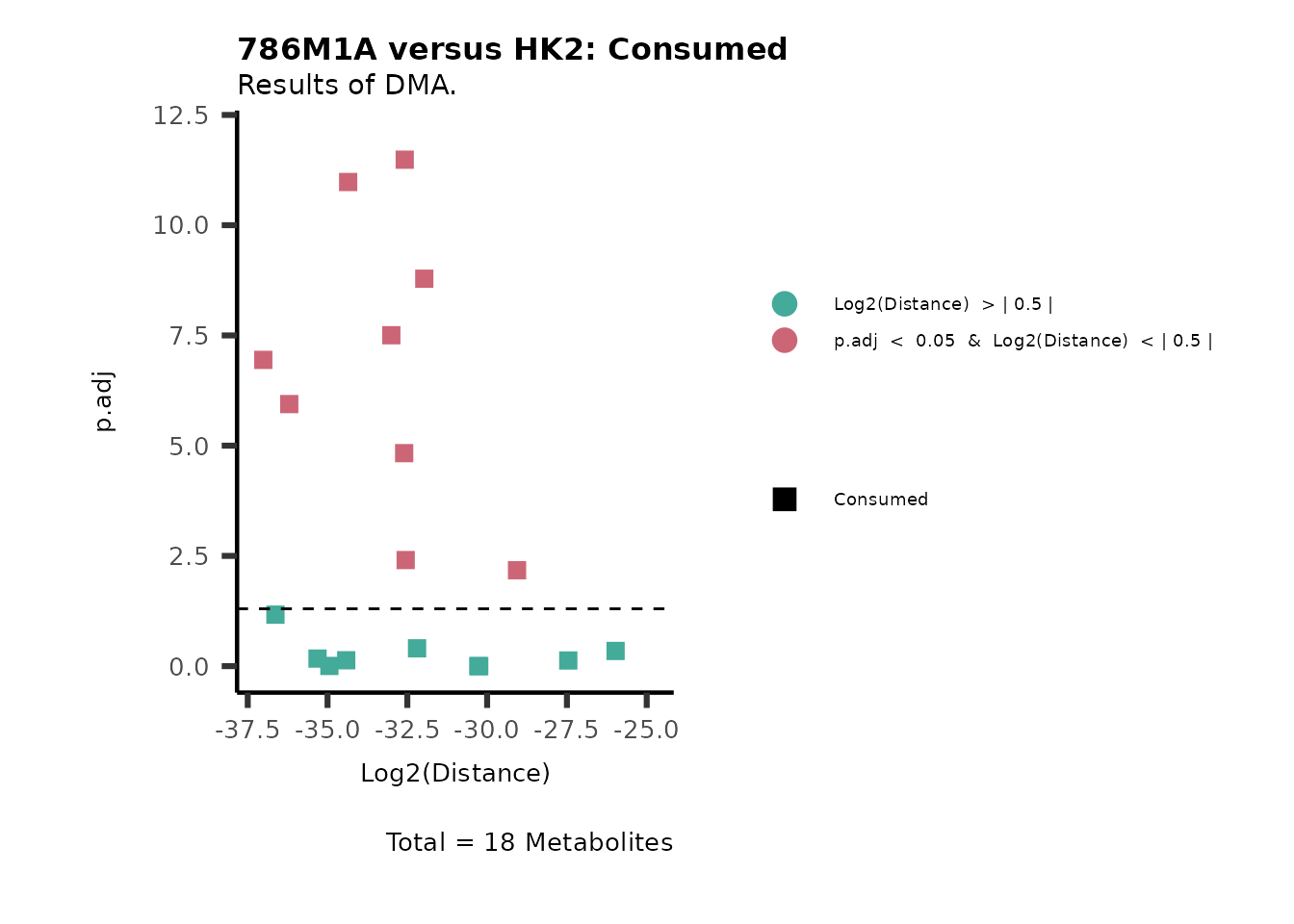
Figure: Standard figure displaying DMA results.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).
#> Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).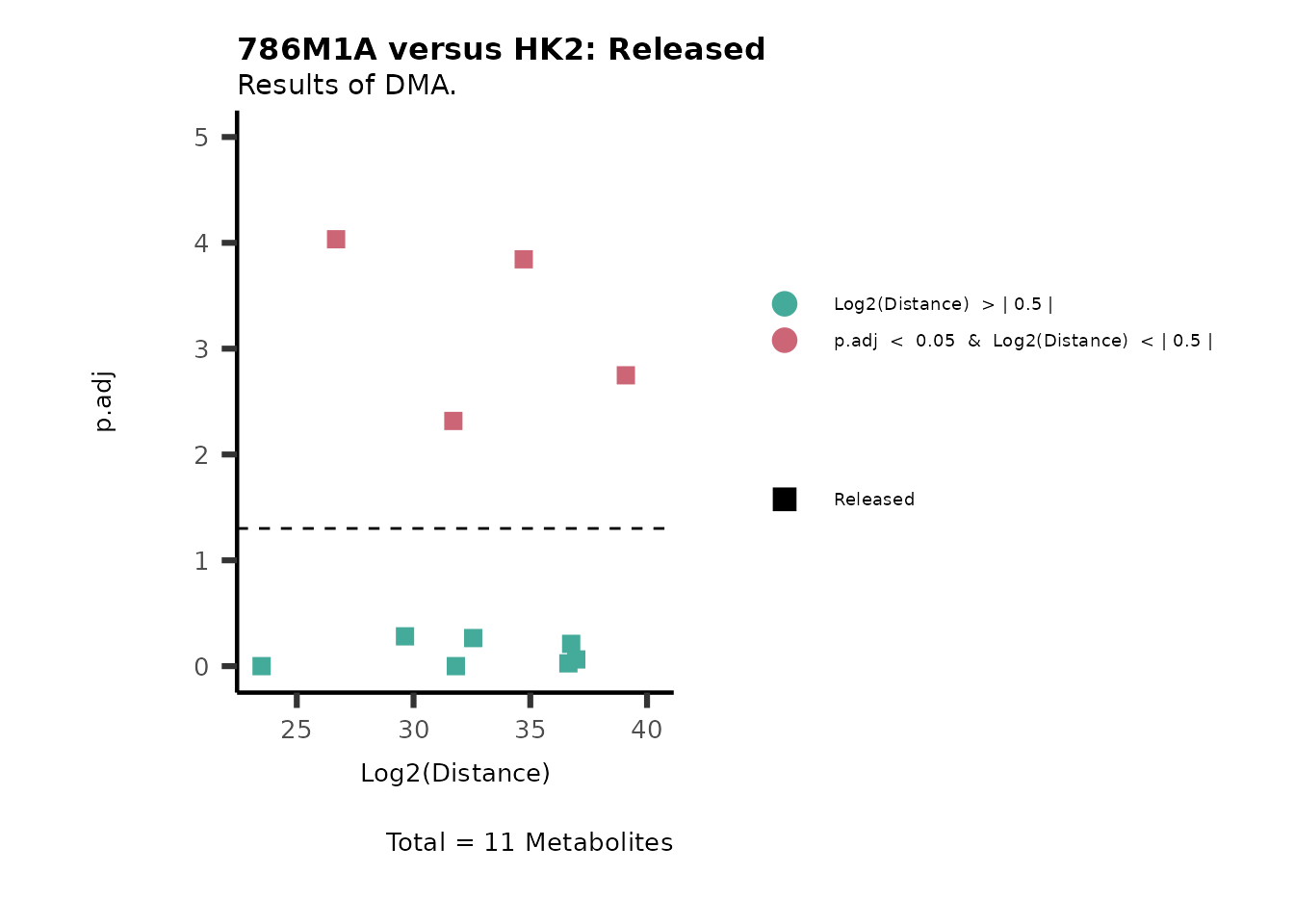
Figure: Standard figure displaying DMA results.
Given that we also know, which metabolic pathway the metabolites
correspond to, we can add this information into the plot. This is also a
good example to showcase the flexibility of the visualisation function:
Either you use the parameter
Plot_SettingsFile= MetaData_Metab as above, but as we have
the column “Pathway” also in our Input_data you can also pass
Plot_SettingsFile= DMA_786-M1A_vs_HK2 or simply use the
default Plot_SettingsFile=NULL, in which case the
Plot_SettingsInfo information (here color)
will be used from Input_data.
#Now we can use color for the pathways and shape for the metabolite clusters:
MetaProViz::VizVolcano(PlotSettings="Standard",
SettingsInfo= c(individual="CoRe", shape="CoRe_specific", color="Pathway"),
SettingsFile= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
x= "Log2(Distance)",
PlotName= "786M1A versus HK2",
Subtitle= "Results of DMA." )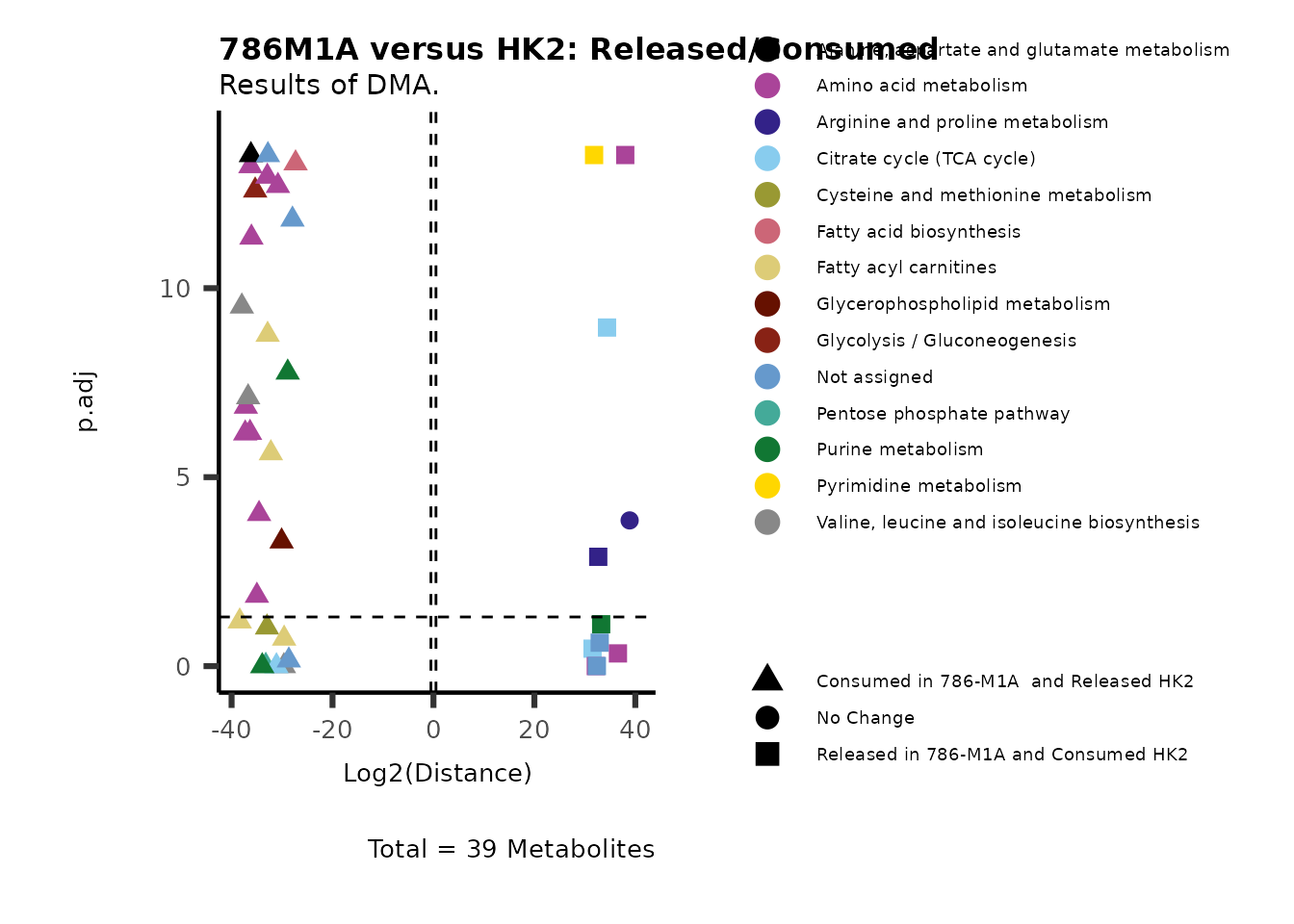
Figure: Standard figure displaying DMA results colour coded for metabolic pathways and shaped for metabolic clusters.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).
#> Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).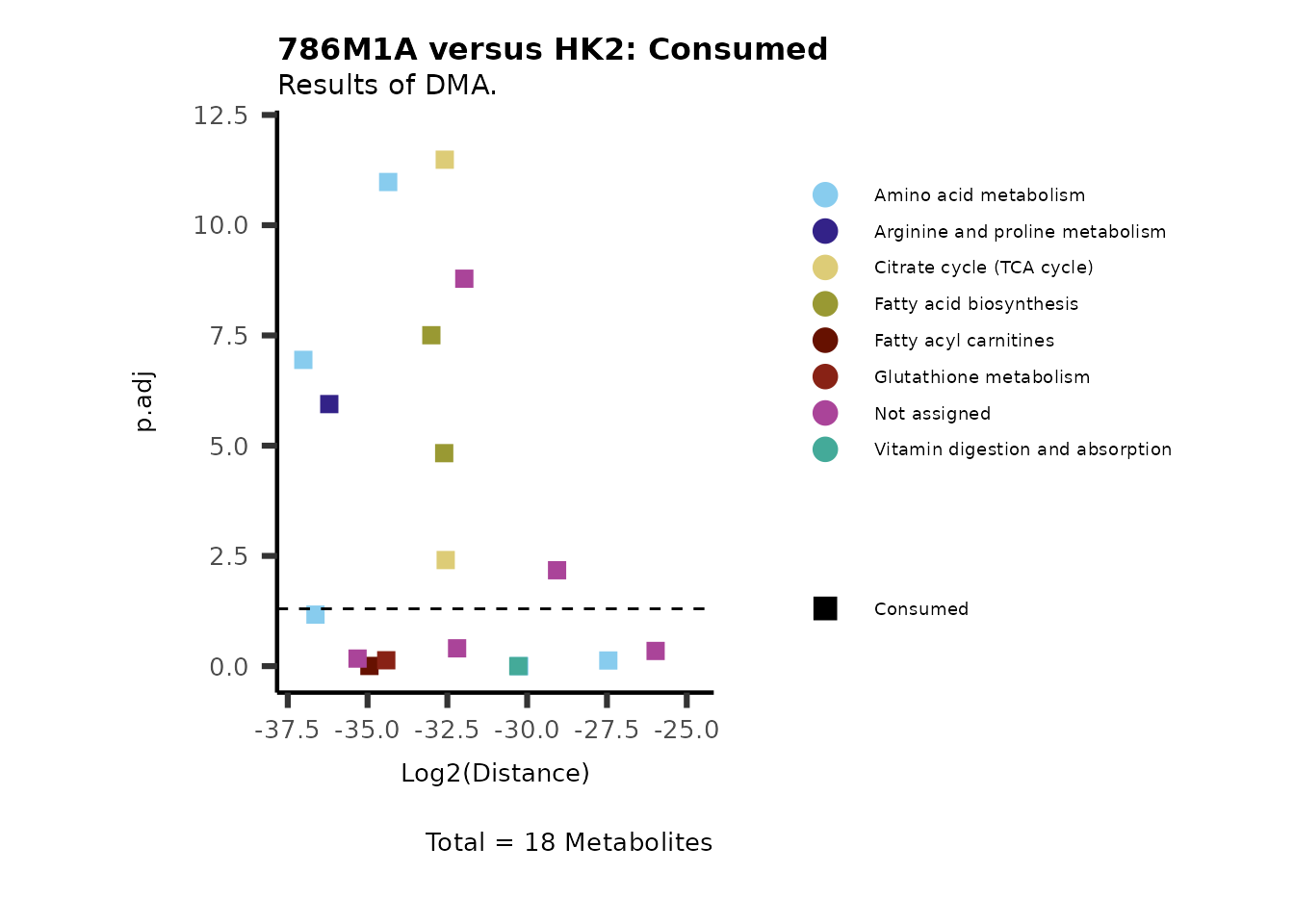
Figure: Standard figure displaying DMA results colour coded for metabolic pathways and shaped for metabolic clusters.
#> Warning: Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).
#> Removed 2 rows containing missing values or values outside the scale range
#> (`geom_vline()`).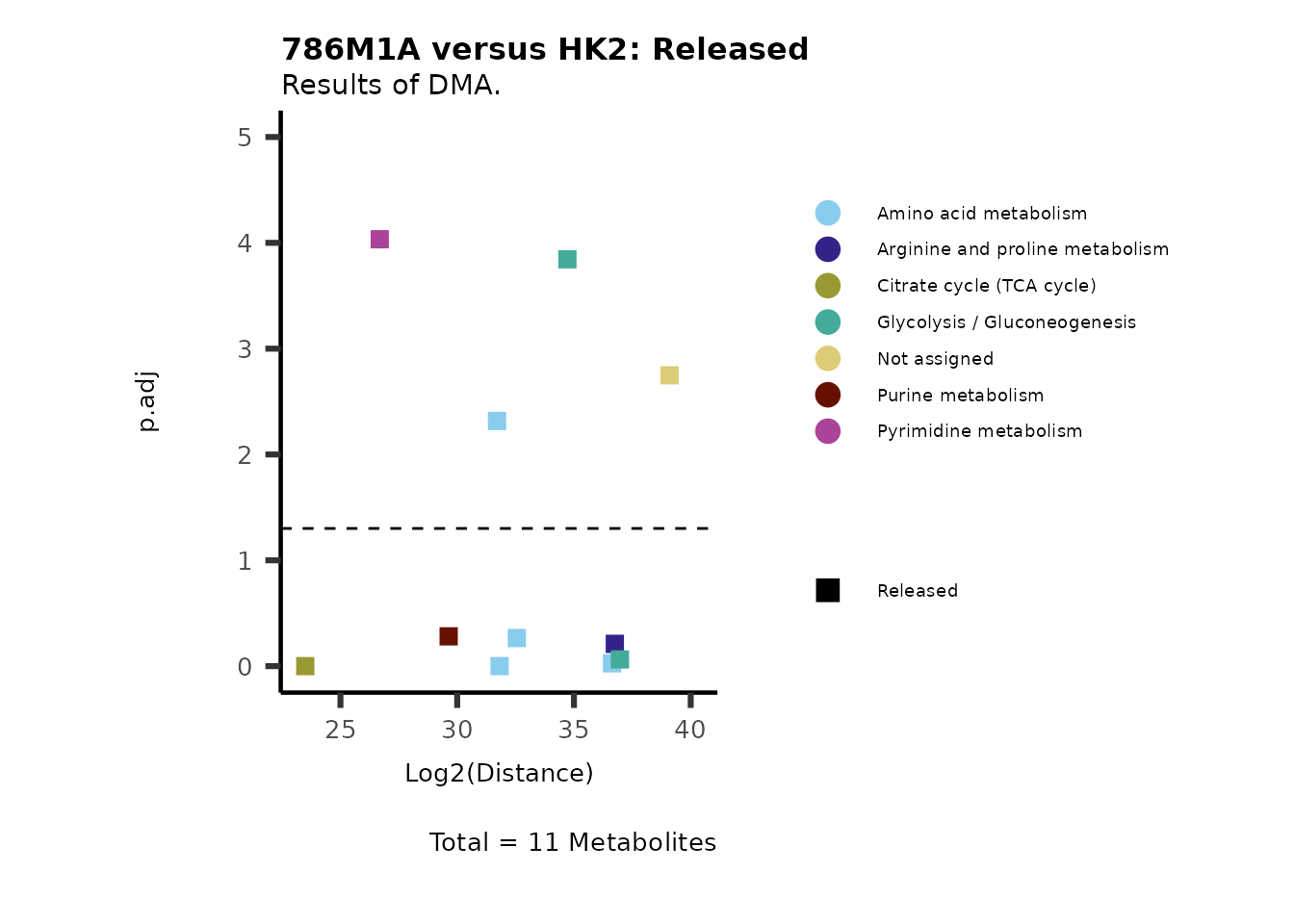
Figure: Standard figure displaying DMA results colour coded for metabolic pathways and shaped for metabolic clusters.
Comparison
The parameter Plot_Settings="Compare" is helpful if you
have performed multiple comparisons and seek to compare two of them in
one plot:
#Make the plot
MetaProViz::VizVolcano(PlotSettings="Compare",
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
InputData2= DMA_Annova[["DMA"]][["786-O_vs_HK2"]]%>%column_to_rownames("Metabolite"),
ComparisonName= c(InputData="786M1A_vs_HK", InputData2= "786-O_vs_HK2"),
x= "Log2(Distance)",
PlotName= "786M1A vs HK2 compared to 7860 vs HK2",
Subtitle= "Results of DMA" )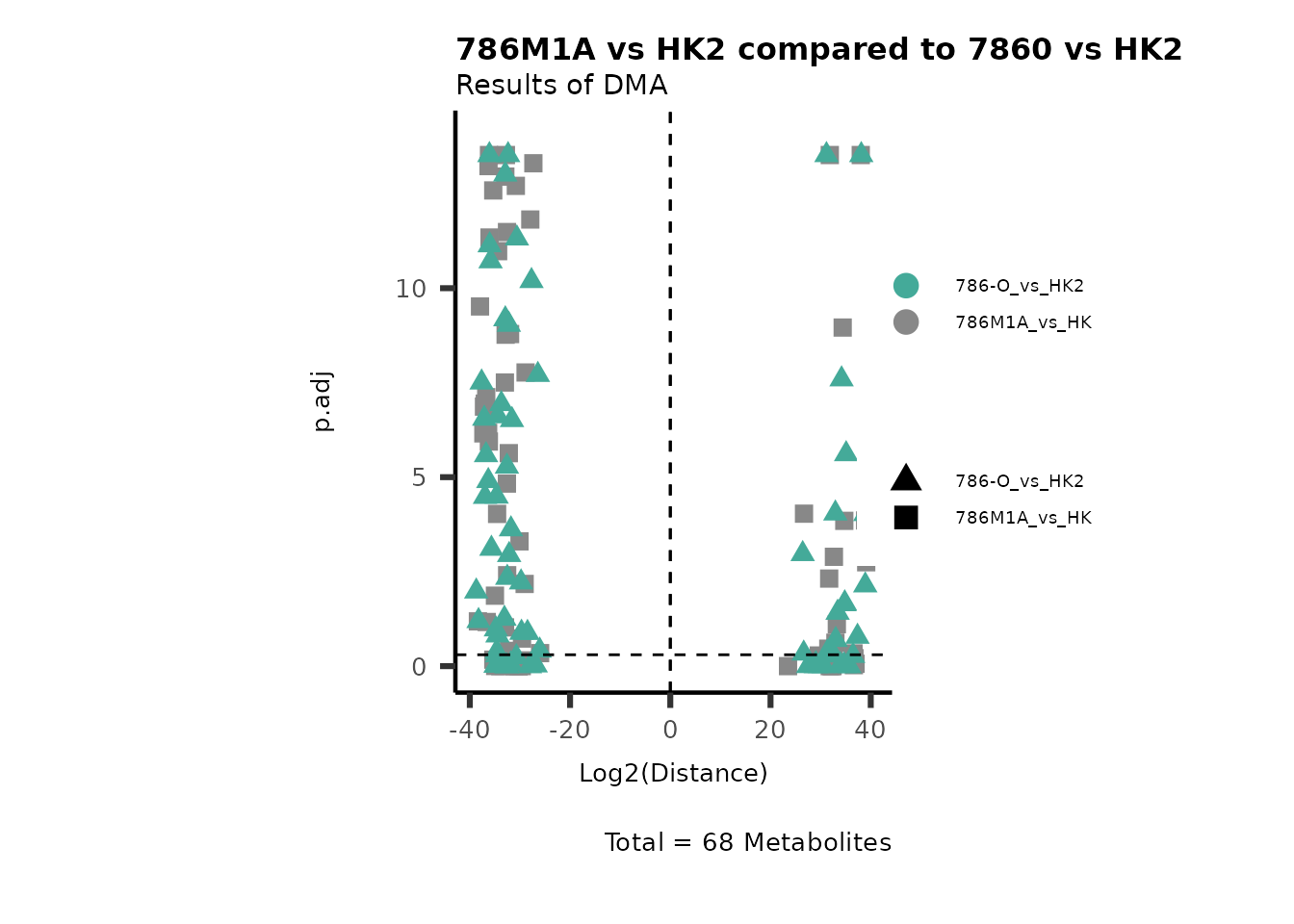
Figure: Comparison.
Of course you have option to use shape or color to further customize
your graph as well as make individual plots:
#Make the plot
MetaProViz::VizVolcano(PlotSettings="Compare",
SettingsInfo= c(color="Pathway"),
SettingsFile_Metab= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
InputData2= DMA_Annova[["DMA"]][["786-O_vs_HK2"]]%>%column_to_rownames("Metabolite"),
ComparisonName= c(InputData="786M1A_vs_HK", InputData2= "786-O_vs_HK2"),
x= "Log2(Distance)",
PlotName= "786M1A vs HK2 compared to 7860 vs HK2",
Subtitle= "Results of DMA" )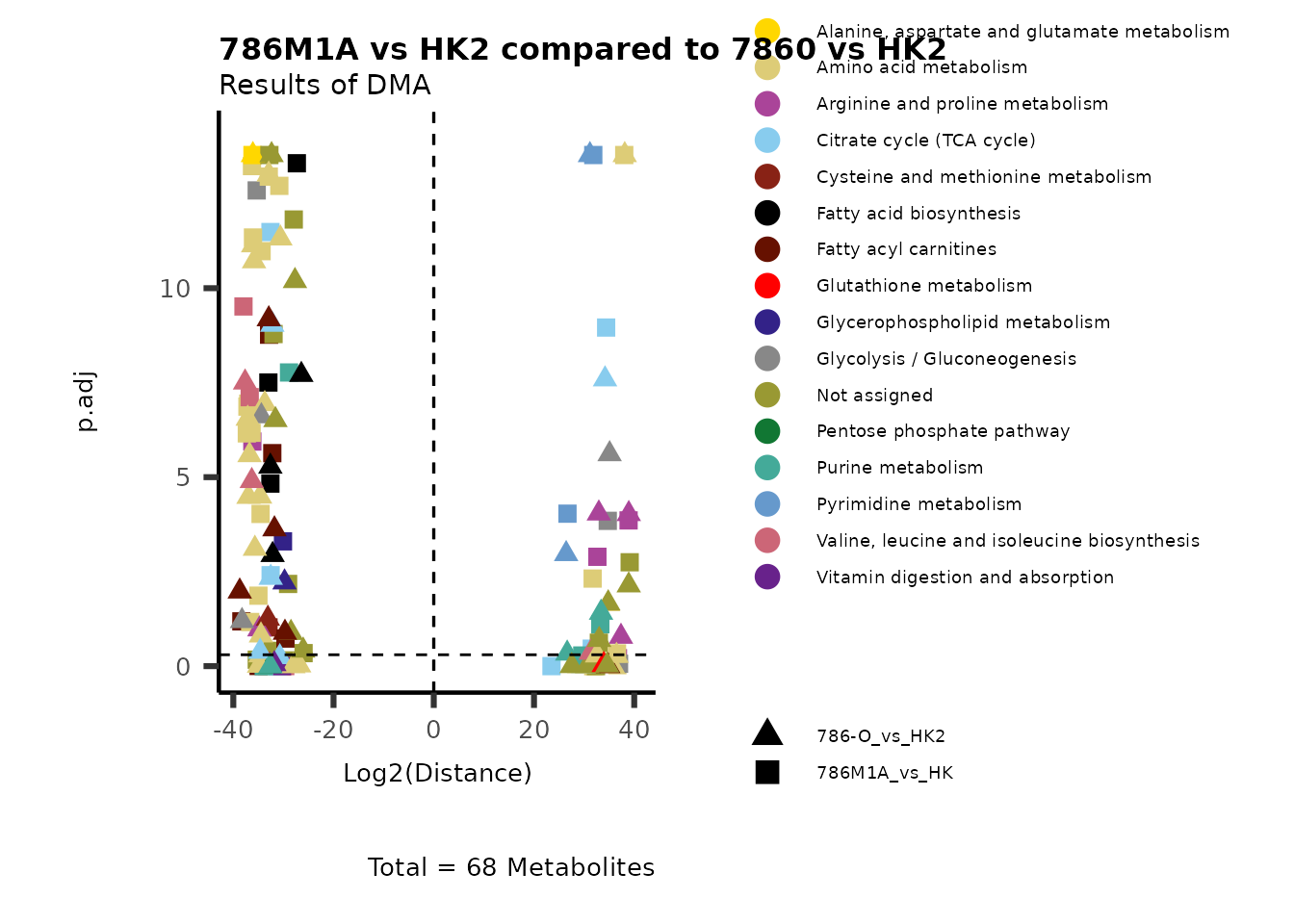
Figure: Comparison.
Now we do individual plots again:
MetaProViz::VizVolcano(PlotSettings="Compare",
SettingsInfo= c(individual="Pathway"),
SettingsFile_Metab= MetaData_Metab,
InputData=DMA_786M1A_vs_HK2%>%column_to_rownames("Metabolite"),
InputData2= DMA_Annova[["DMA"]][["786-O_vs_HK2"]]%>%column_to_rownames("Metabolite"),
ComparisonName= c(InputData="786M1A_vs_HK", InputData2= "786-O_vs_HK2"),
x= "Log2(Distance)",
PlotName= "786M1A vs HK2 compared to 7860 vs HK2",
Subtitle= "Results of DMA" )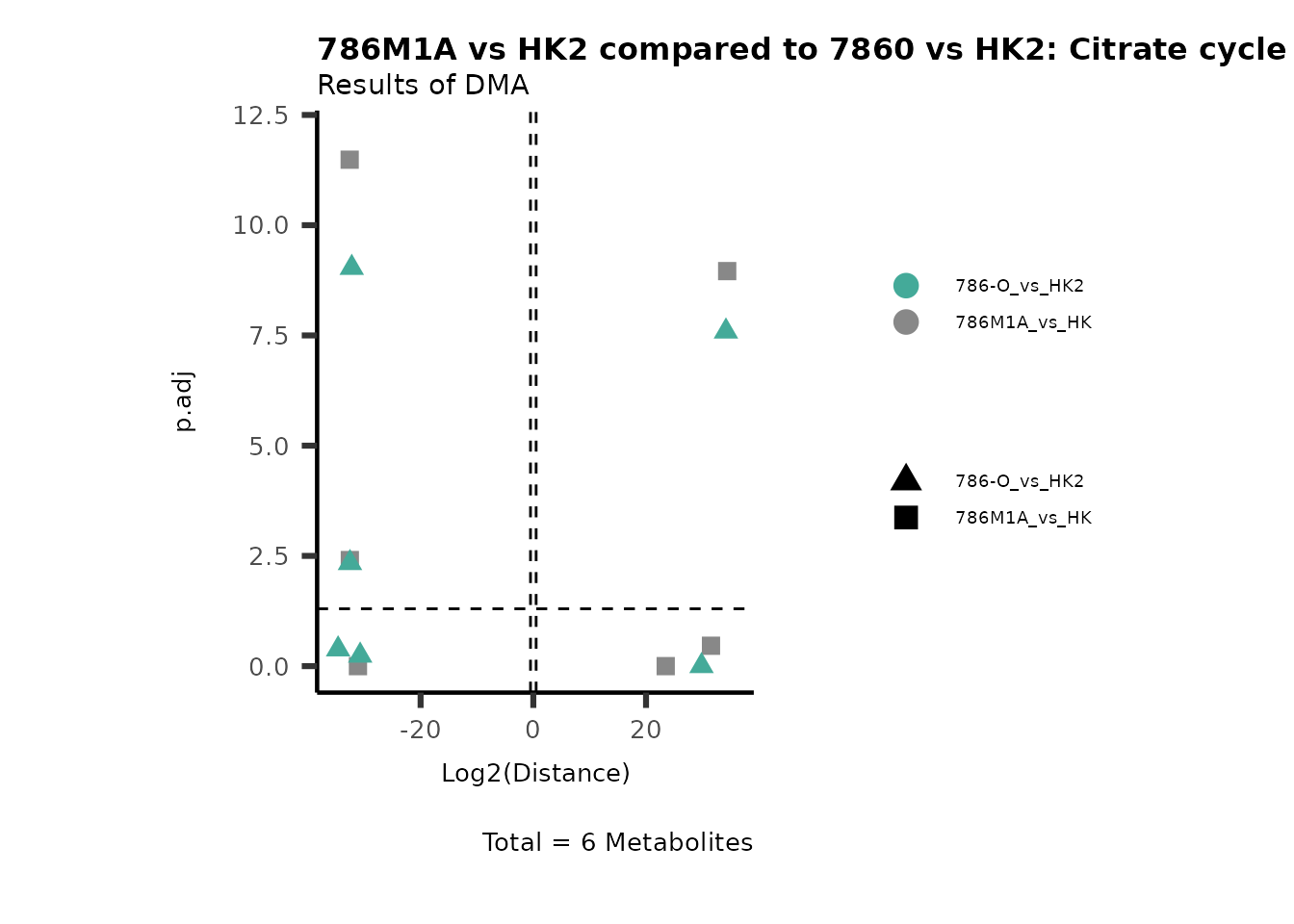
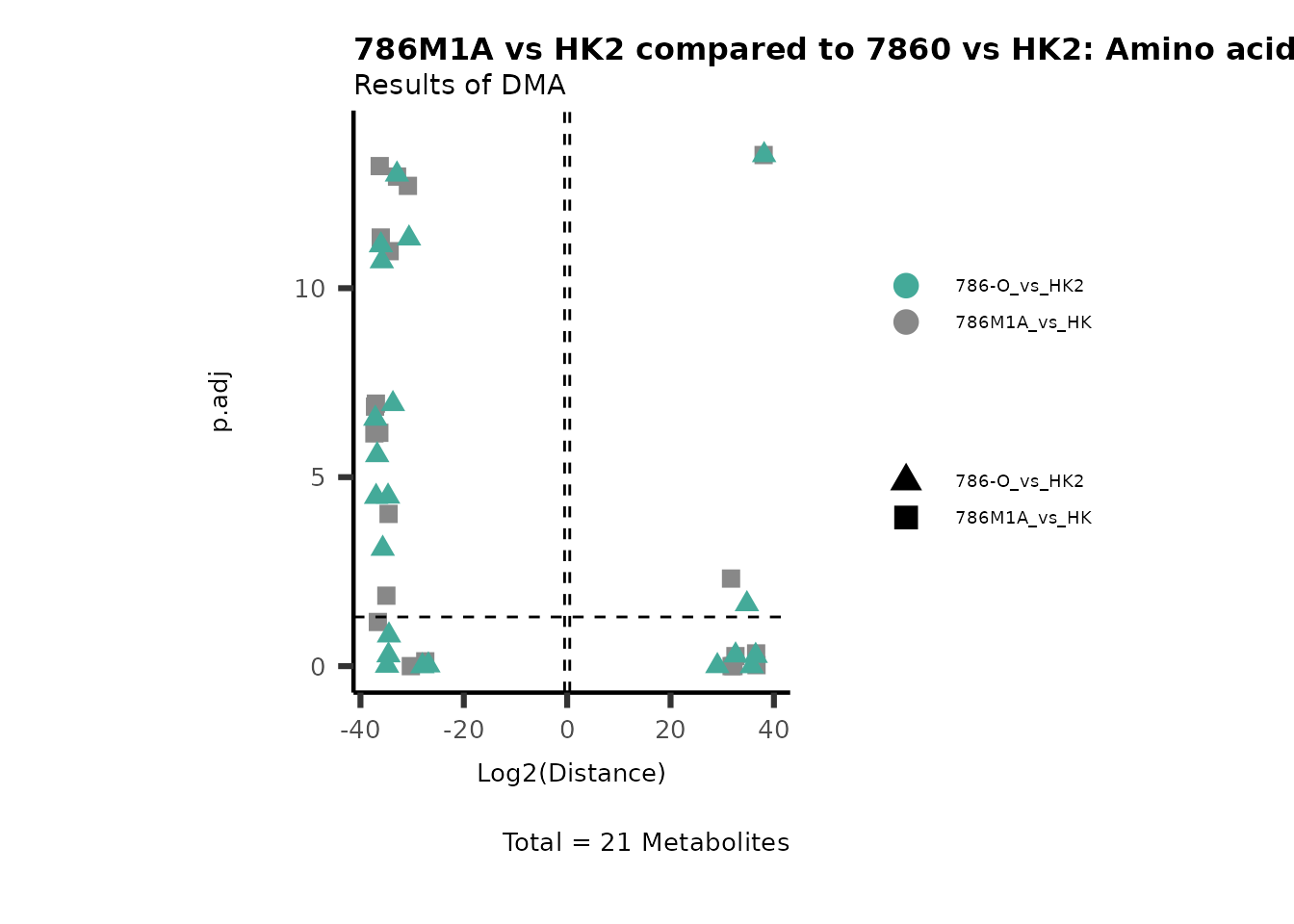

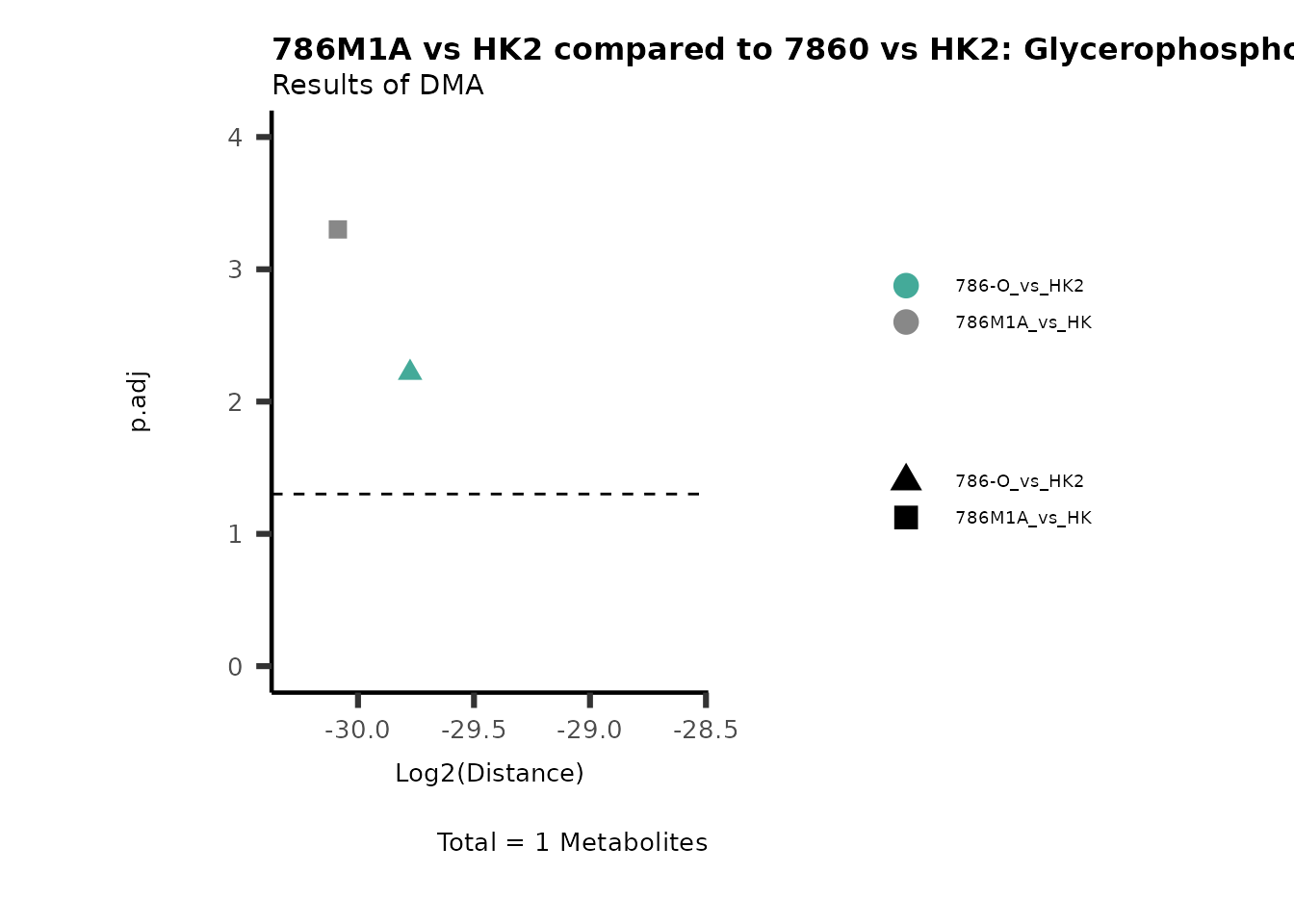
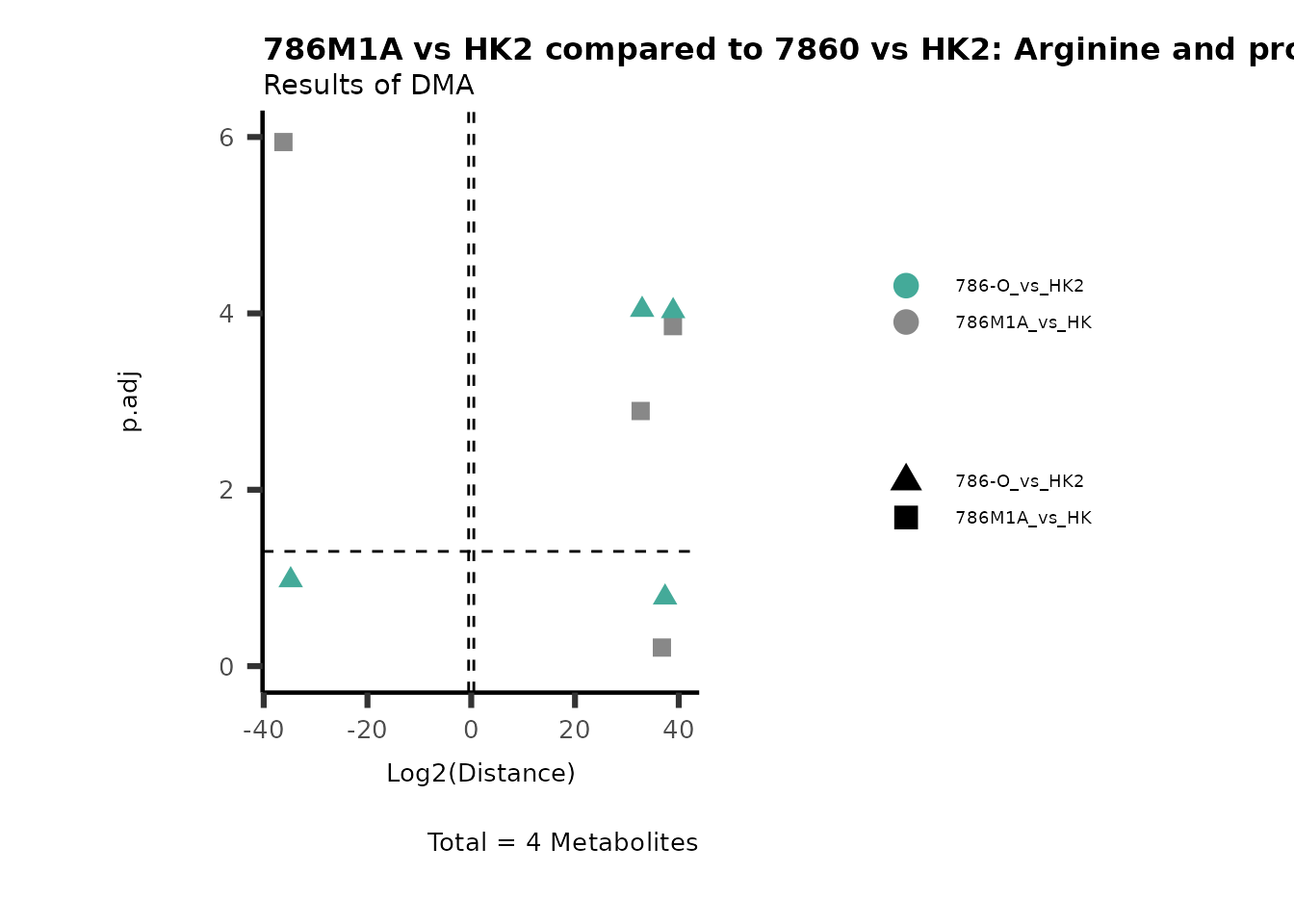

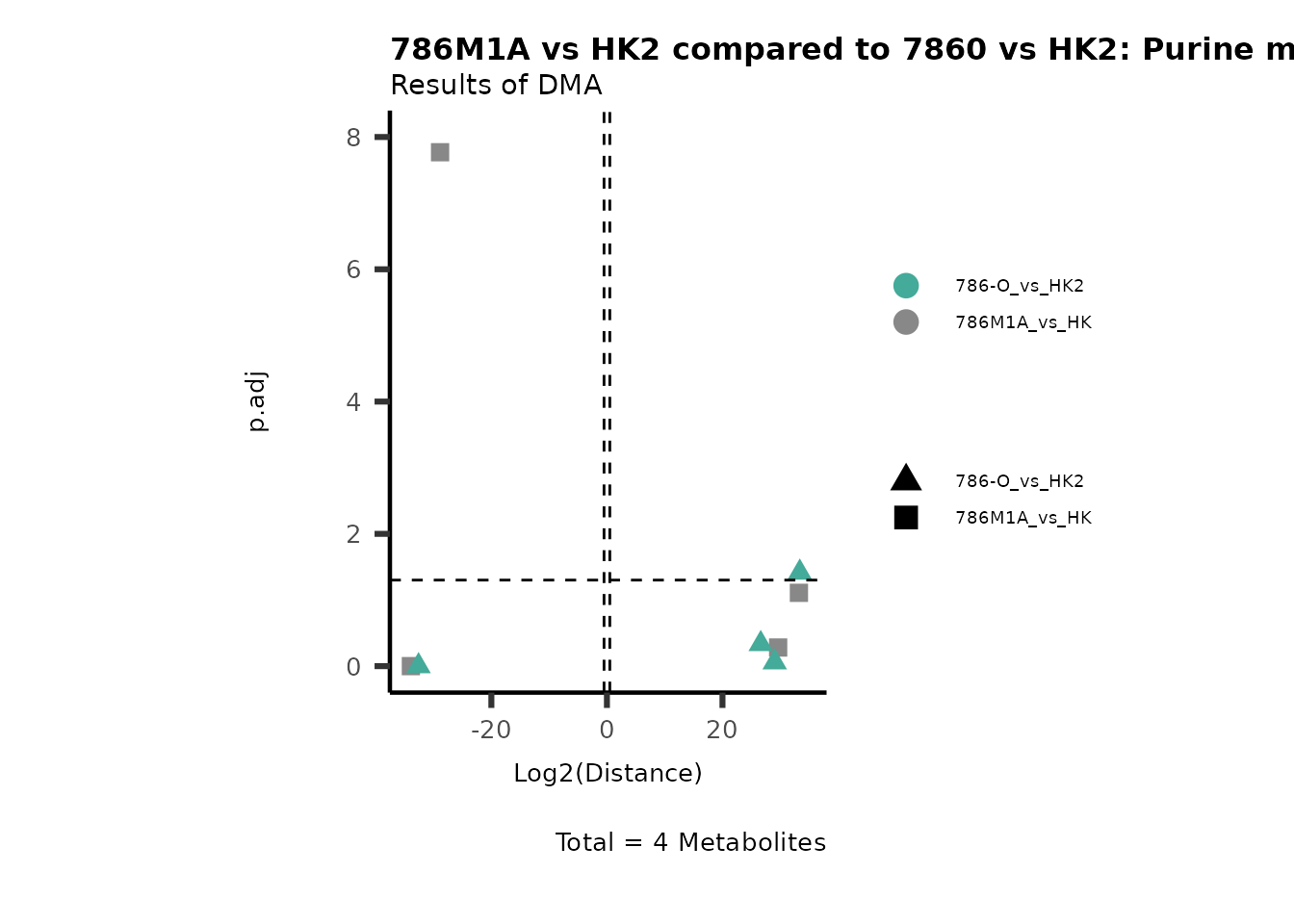

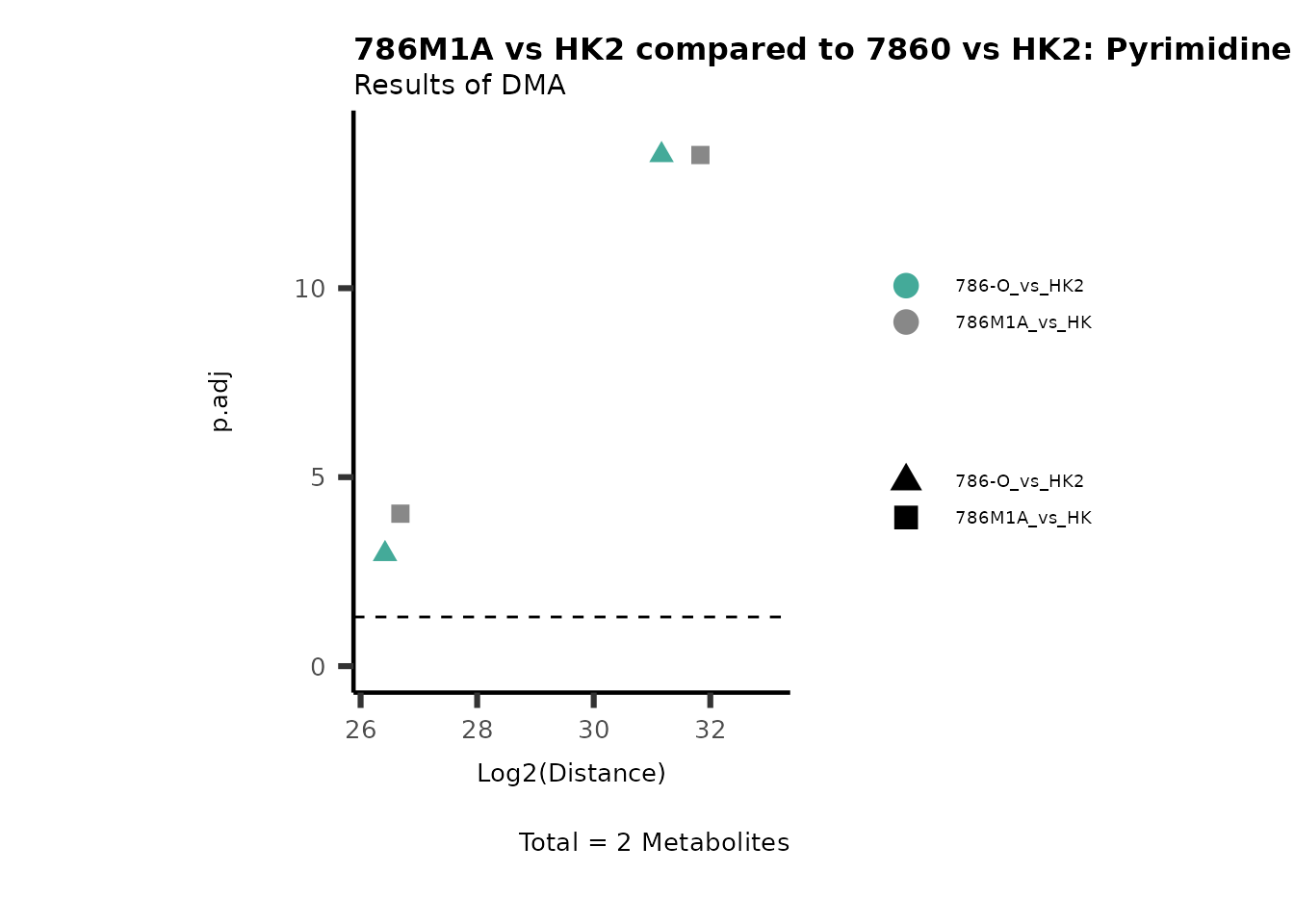
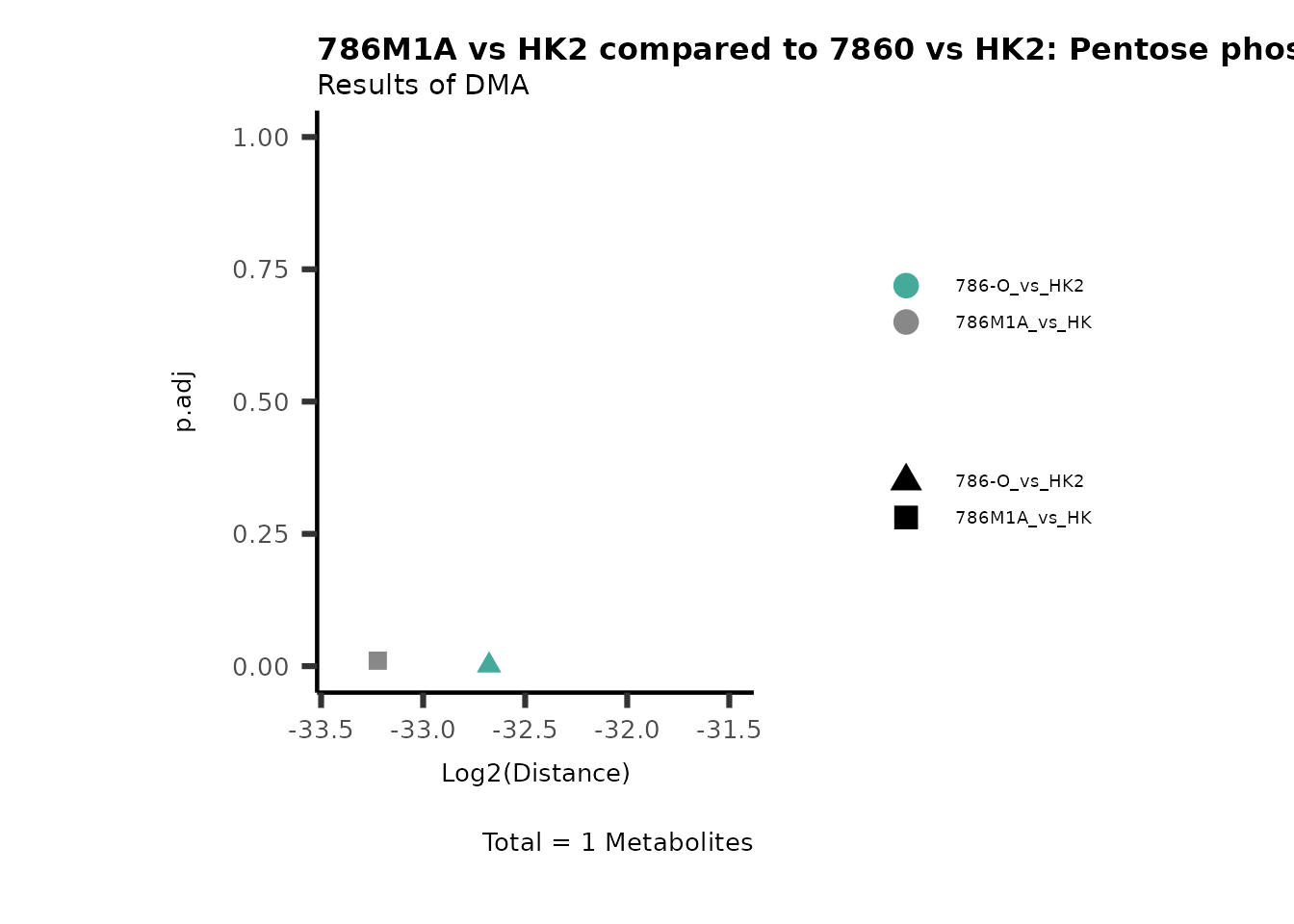
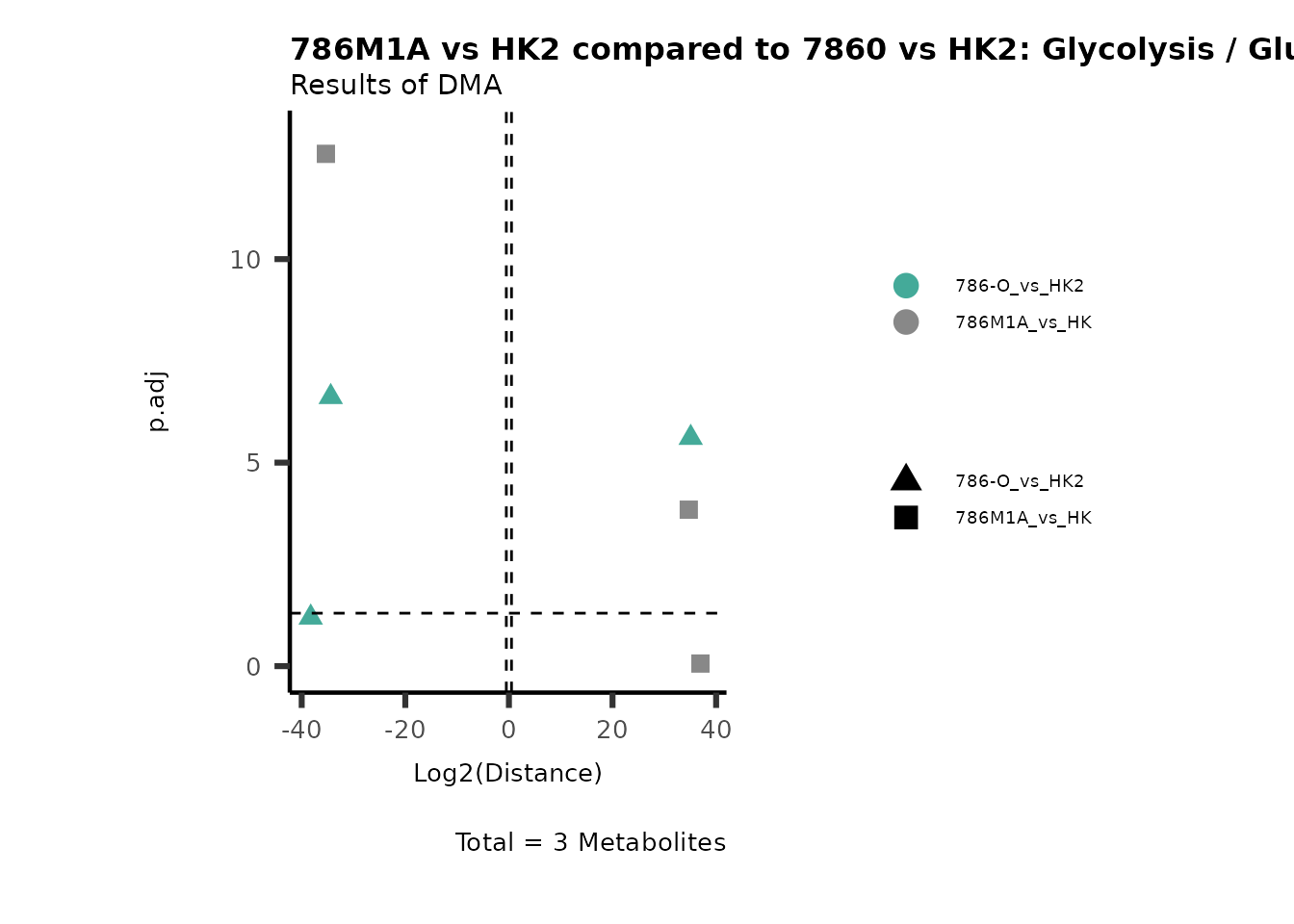
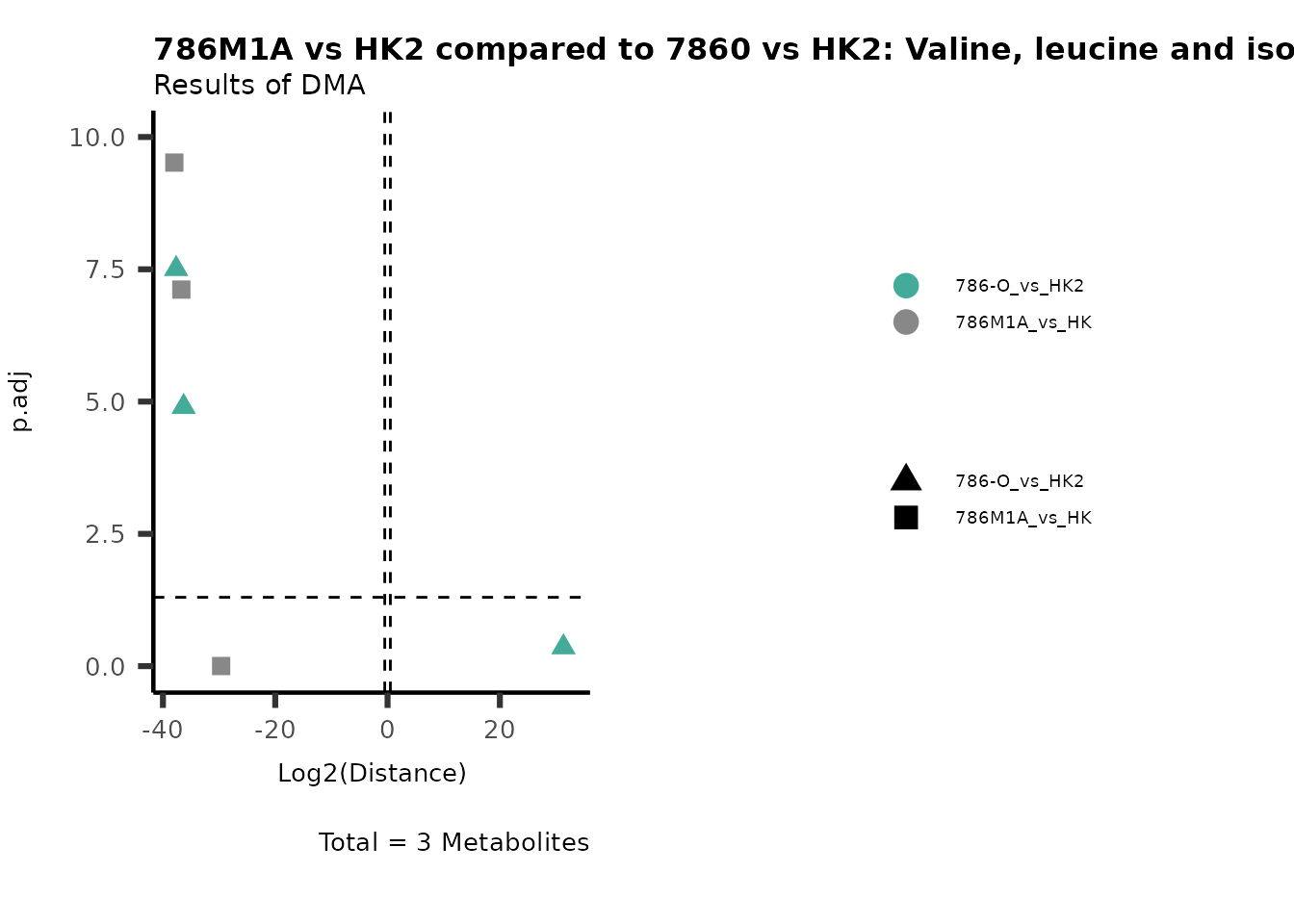
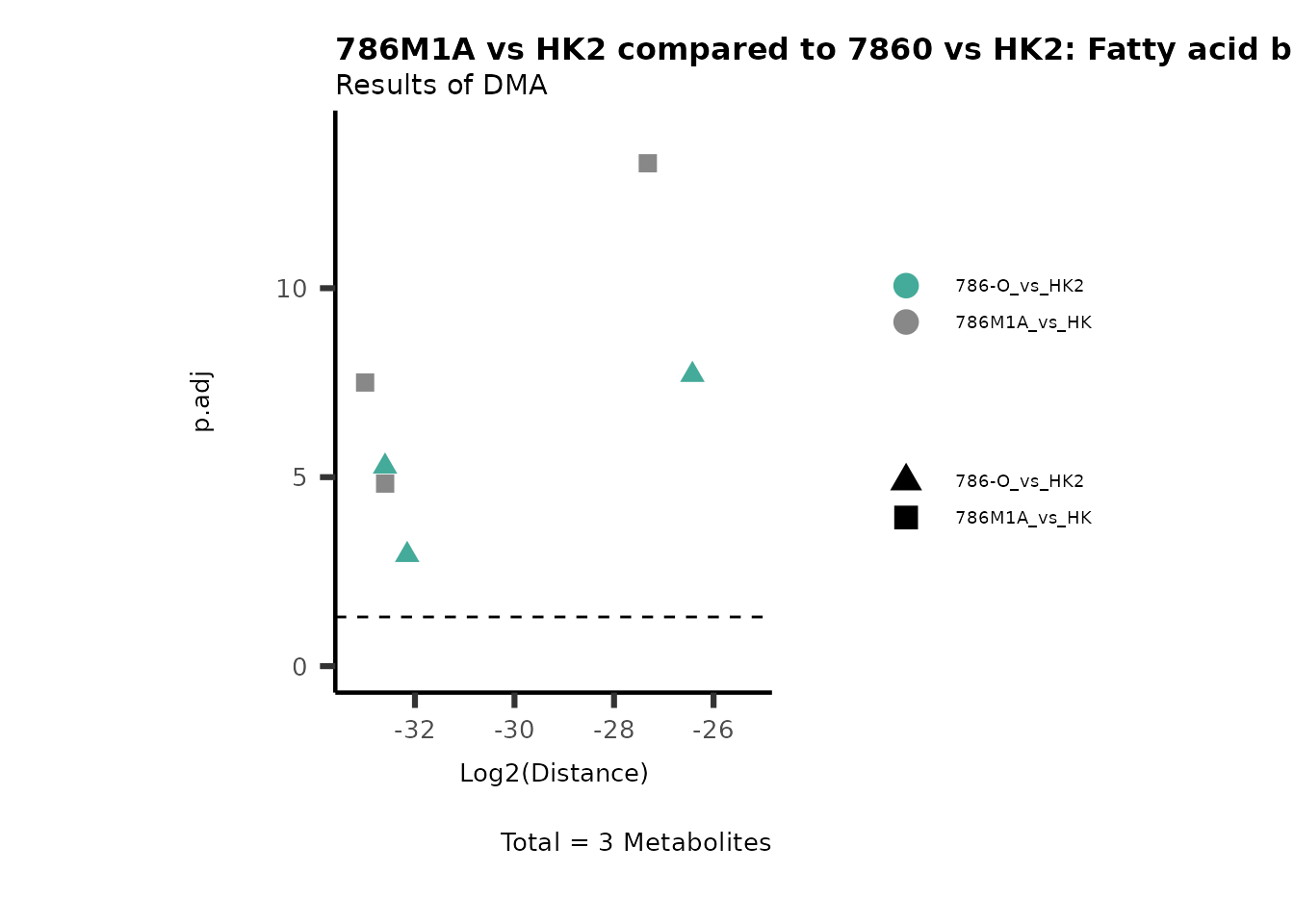
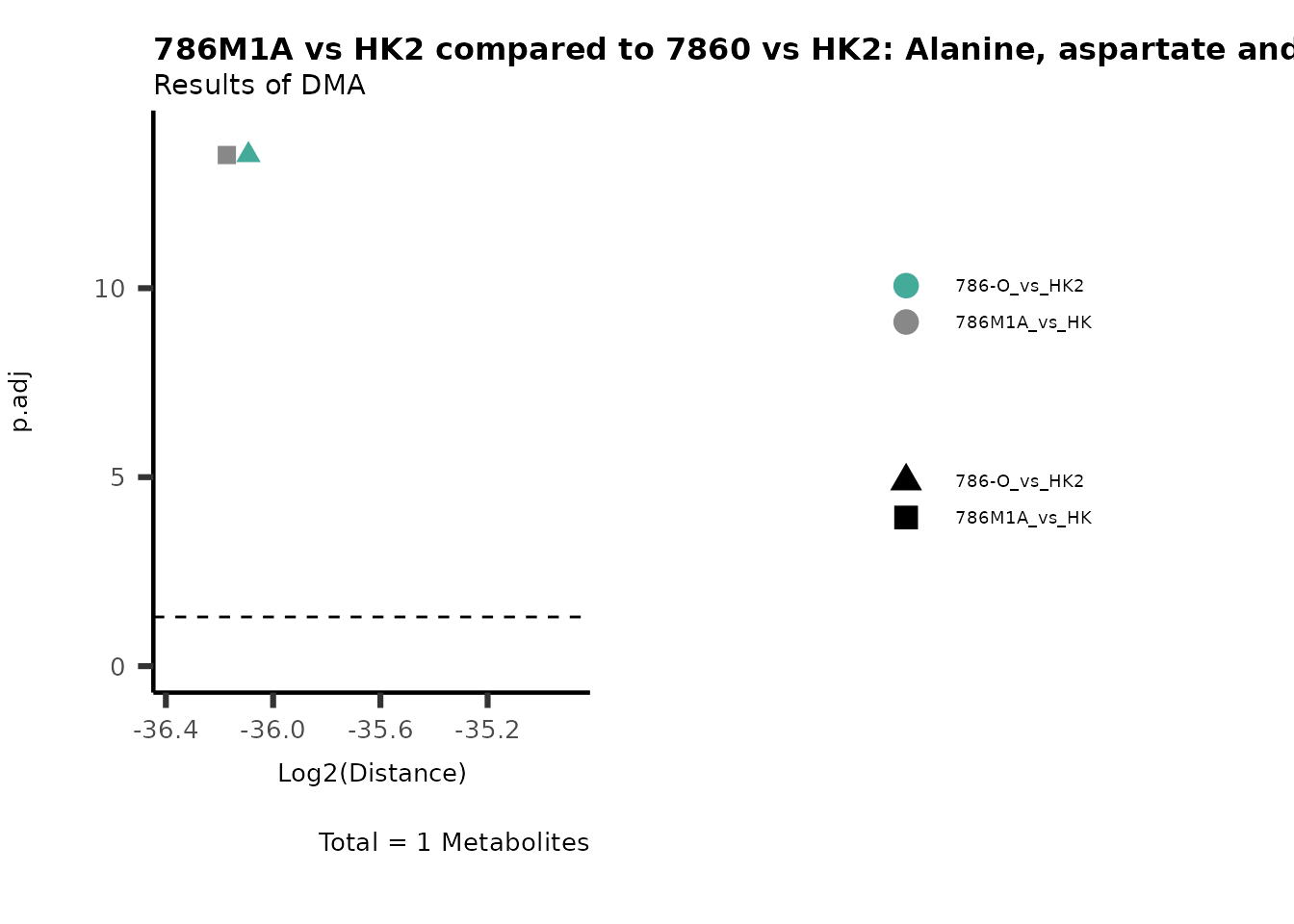
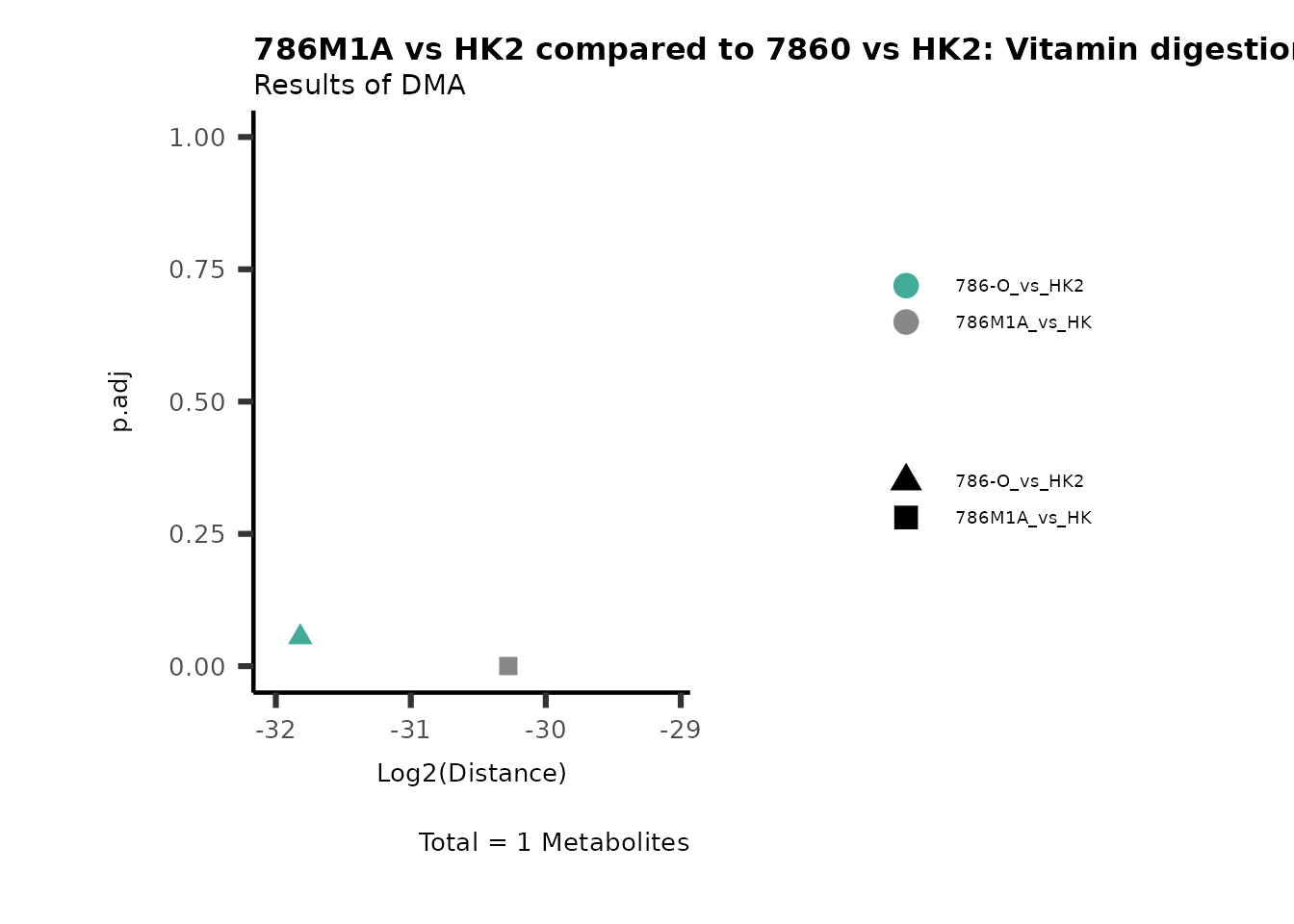
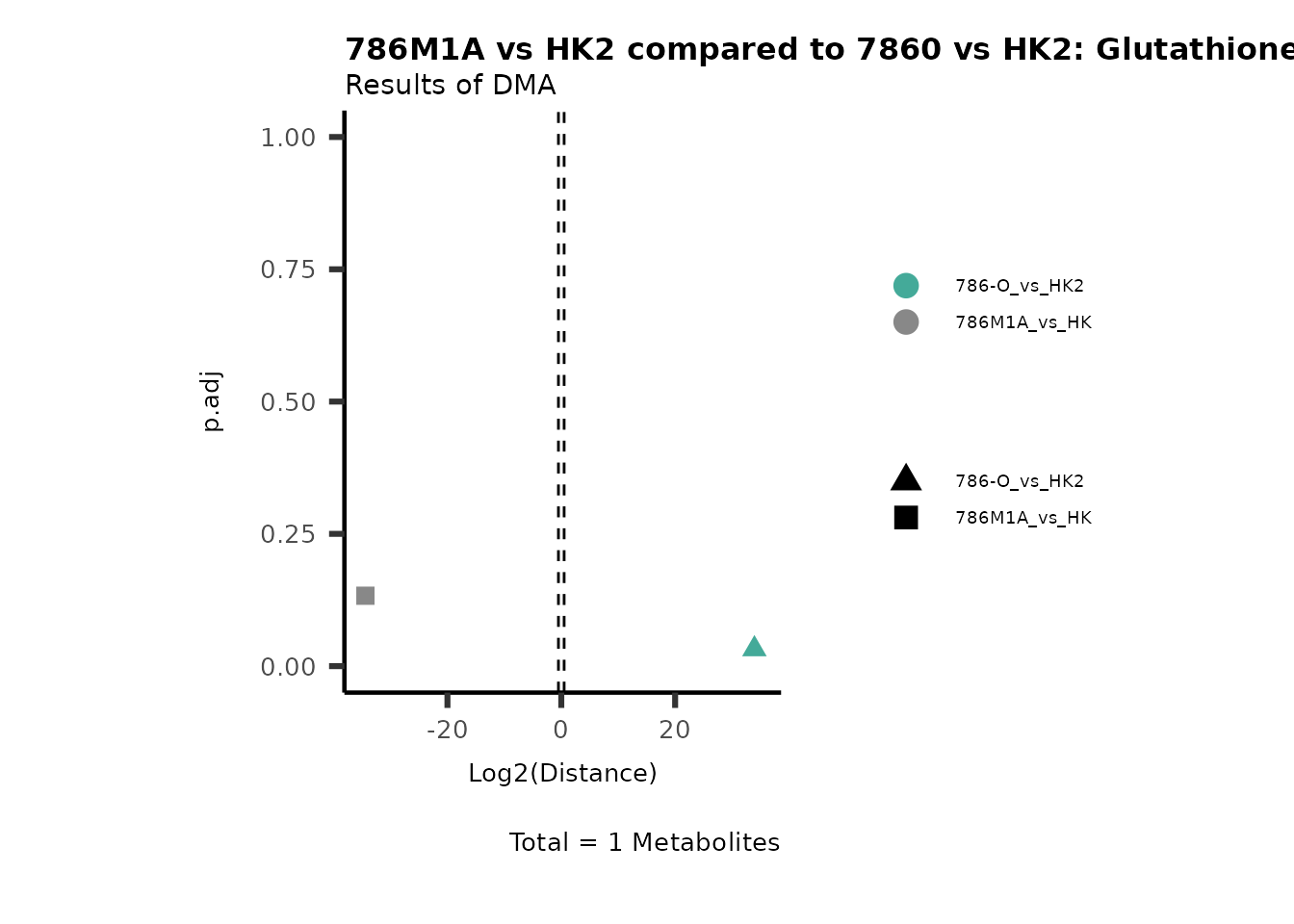
PathwayEnrichmentAnalysis
If you have performed Pathway Enrichment Analysis (PEA) such as ORA
or GSEA, we can also plot the results and add the information into the
Figure legends.
Here we can for example use the results of the ORA we have performed on
the differential expression results. Indeed for DMA_786M1A_vs_HK2 we
have performed ORA on each cluster (consumed, released,
consumed/released). Here, I will plot the ORA results of the metabolites
that are released in both conditions, HK2 and 786-M1A.
#Prepare the Input:
#1. InputData=Pathway analysis input: Must have features as column names. Those feature names need to match features in the pathway analysis file SettingsFile_Metab.
InputPEA <- DMA_786M1A_vs_HK2 %>%
filter(!is.na(KEGGCompound)) %>%
column_to_rownames("KEGGCompound")
#2. InputData2=Pathway analysis output: Must have same column names as SettingsFile_Metab for Pathway name
InputPEA2 <- MC_ORA_786M1A_vs_HK2_Consumed %>%
dplyr::rename("term"="ID")
#3. SettingsFile_Metab= Pathways used for pathway analysis: Must have same column names as SettingsFile_Metab for Pathway name and feature names need to match features in the InputData. PEA_Feature passes this column name!
MetaProViz::VizVolcano(PlotSettings="PEA",
SettingsInfo= c(PEA_Pathway="term",# Needs to be the same in both, SettingsFile_Metab and InputData2.
PEA_stat="p.adjust",#Column InputData2
PEA_score="GeneRatio",#Column InputData2
PEA_Feature="Metabolite"),# Column SettingsFile_Metab (needs to be the same as row names in InputData)
SettingsFile_Metab= KEGG_Pathways,#Must be the pathways used for pathway analysis
InputData= InputPEA, #Must be the data you have used as an input for the pathway analysis
InputData2= InputPEA2, #Must be the results of the pathway analysis
x= "Log2(Distance)",
PlotName= "KEGG",
Subtitle= "PEA" ,
SelectLab = NULL)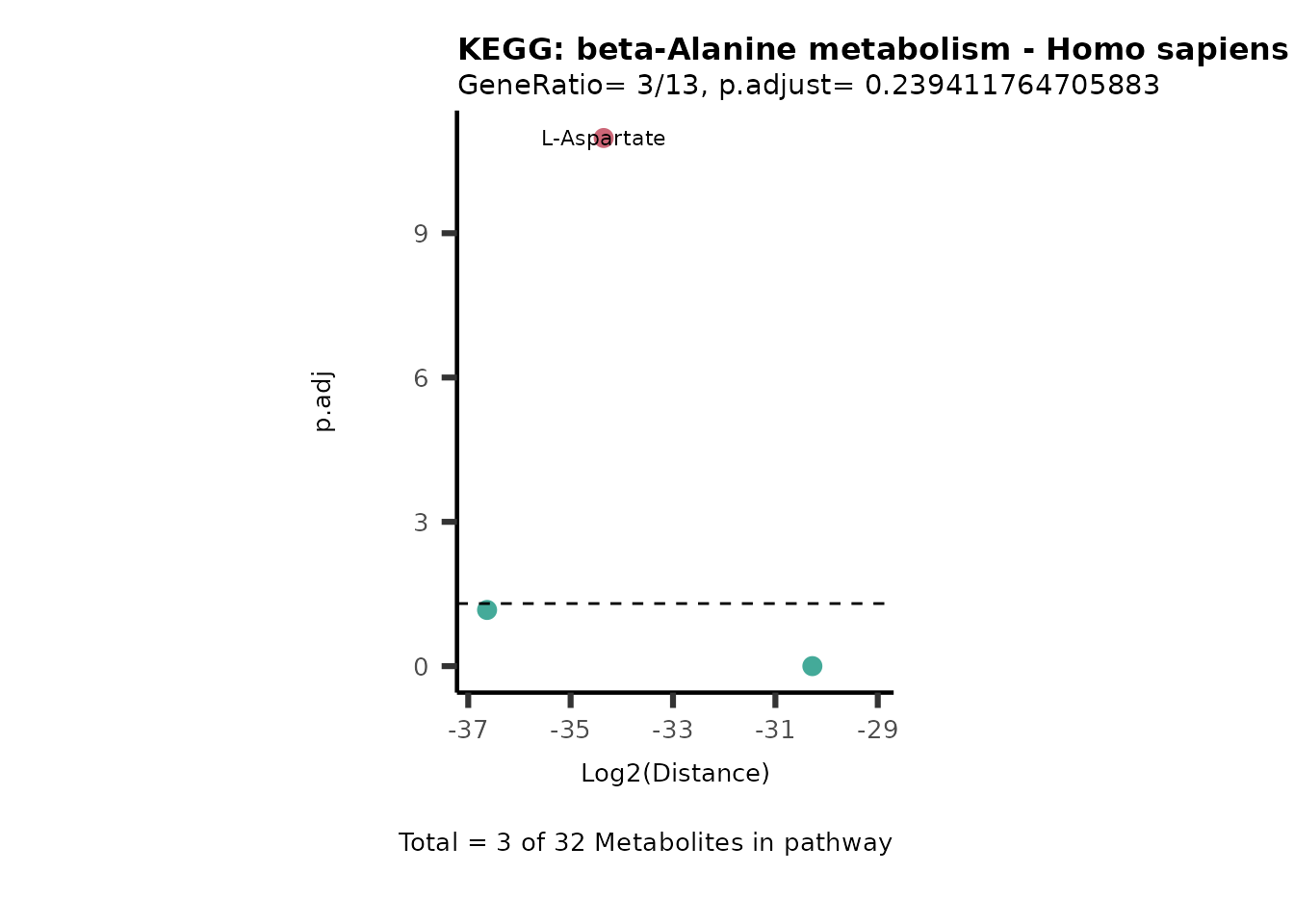

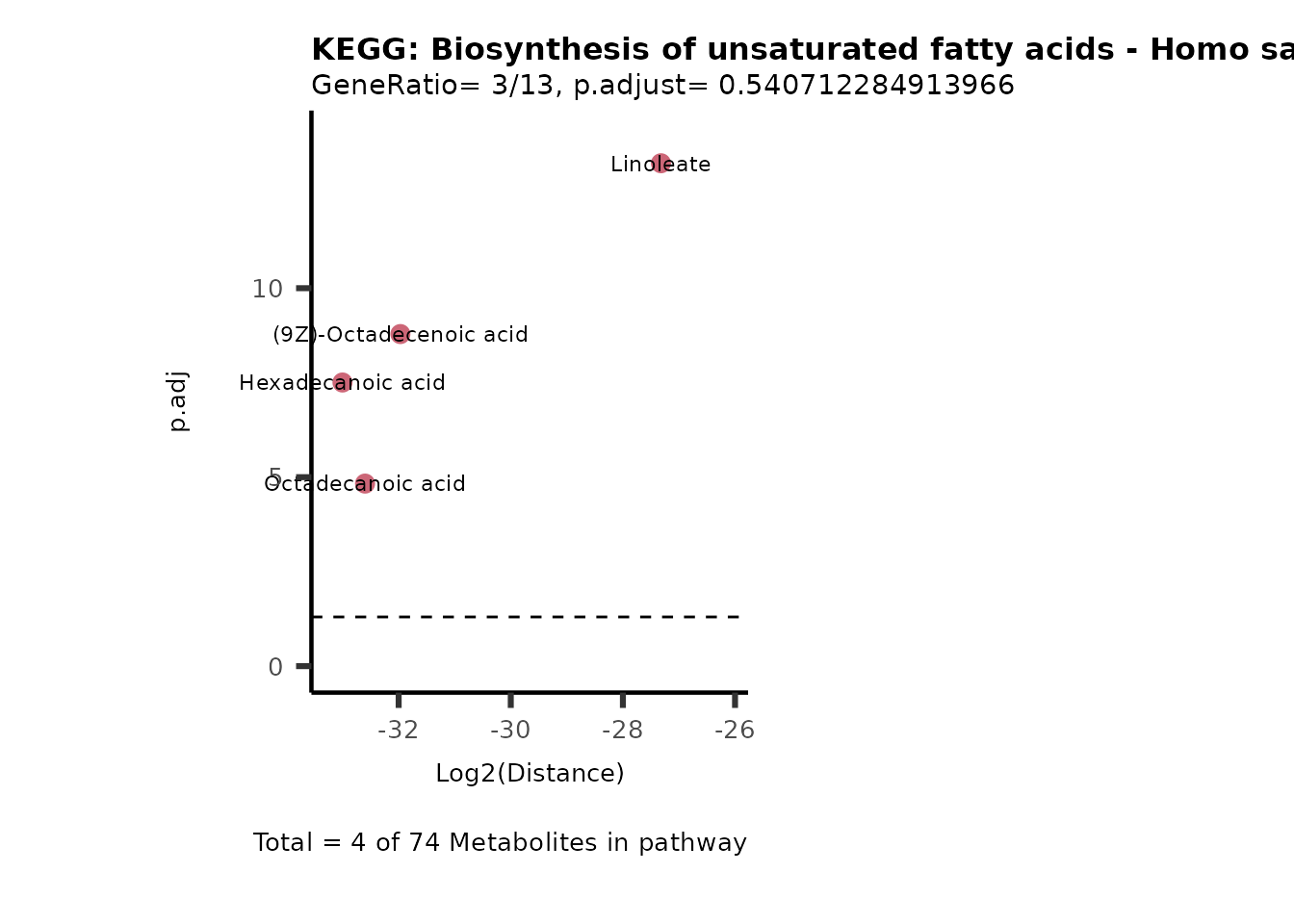
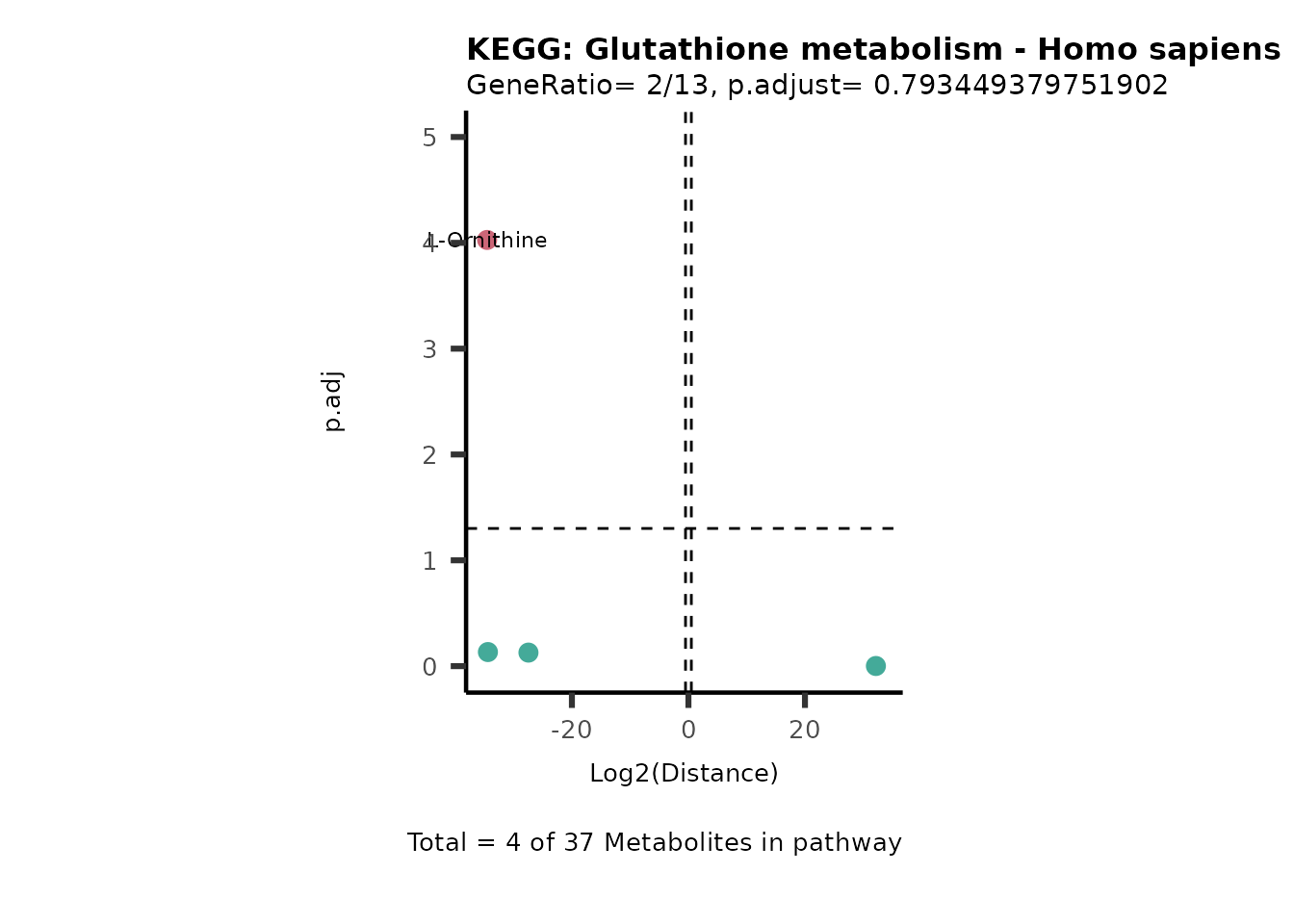
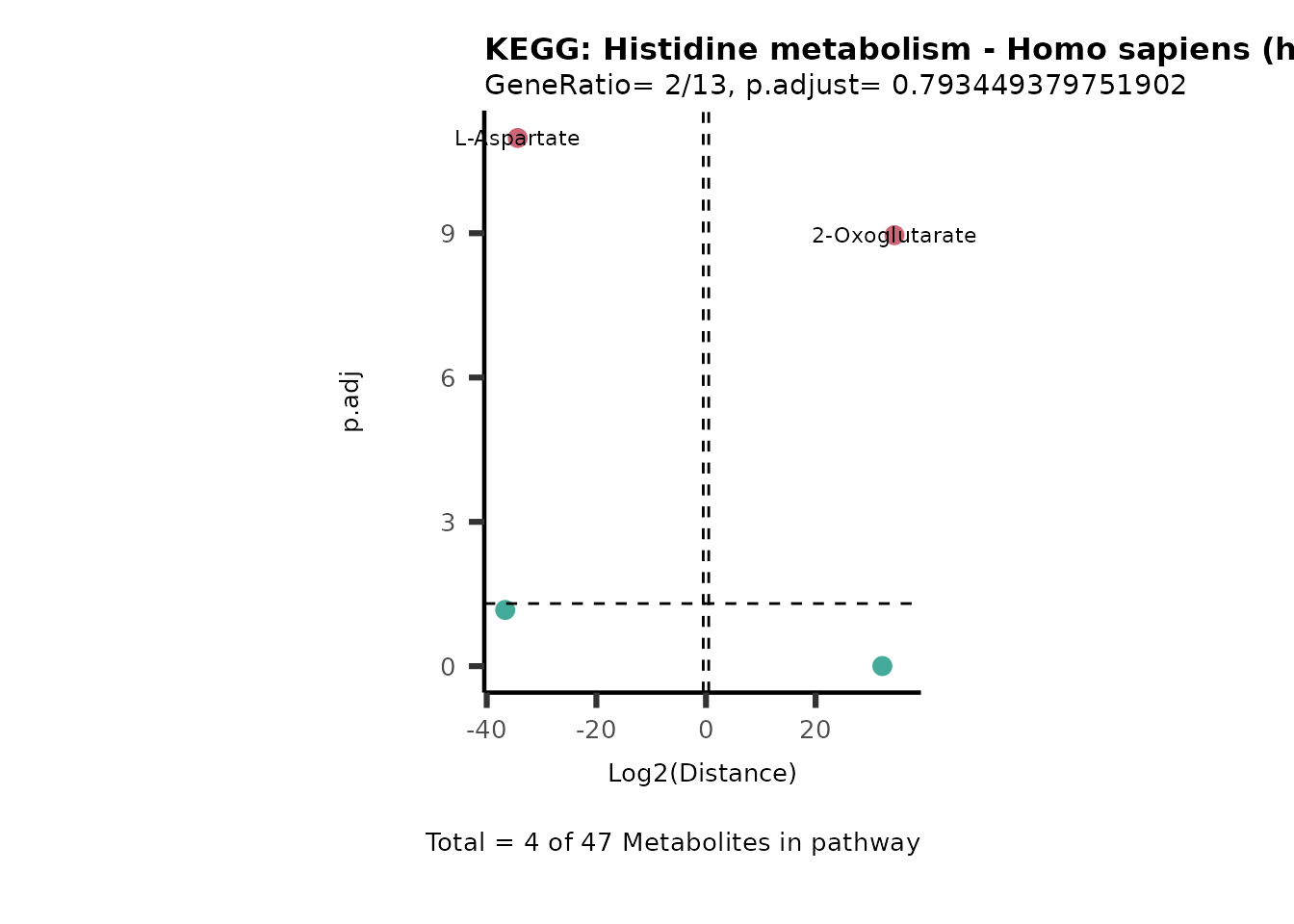
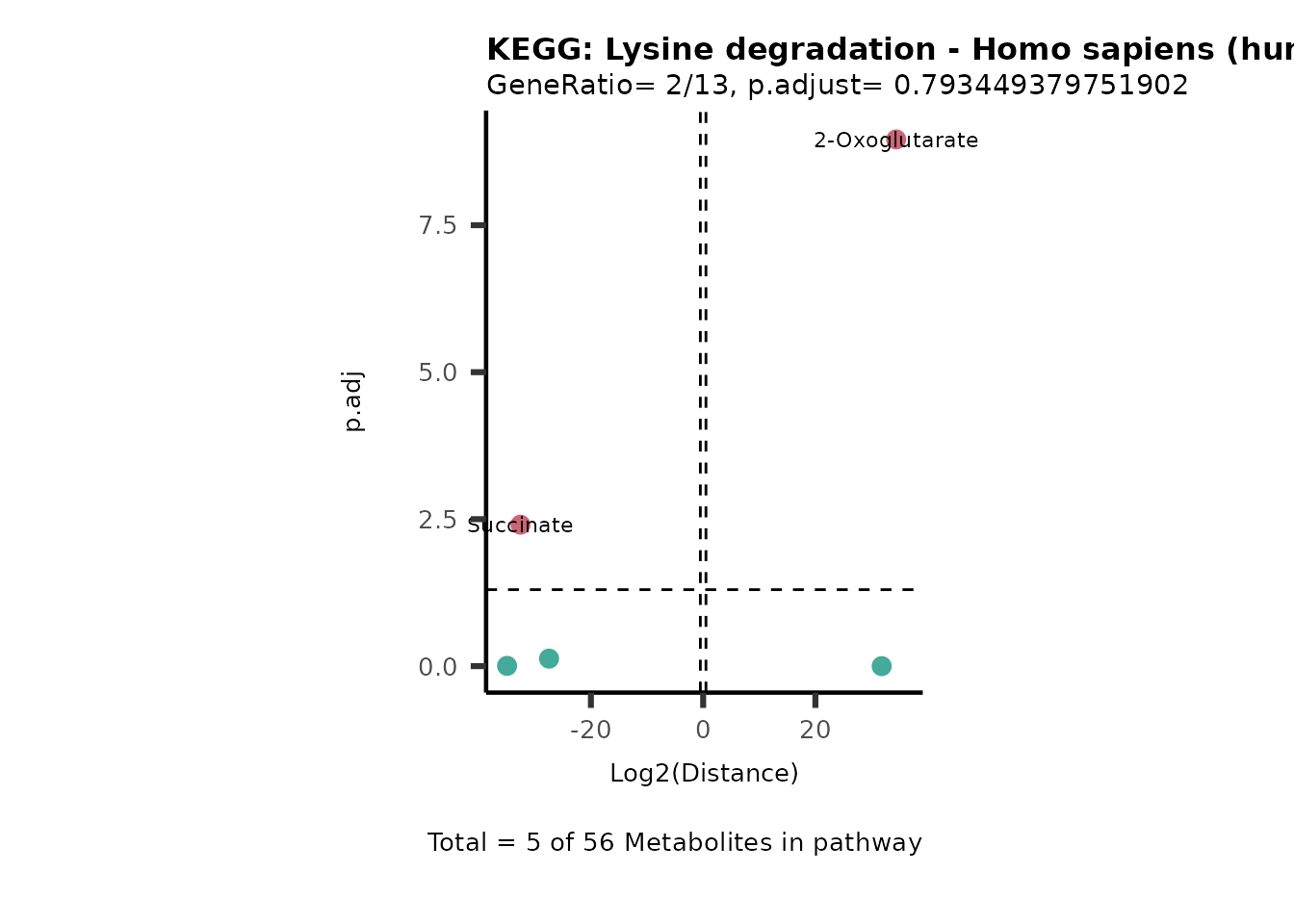
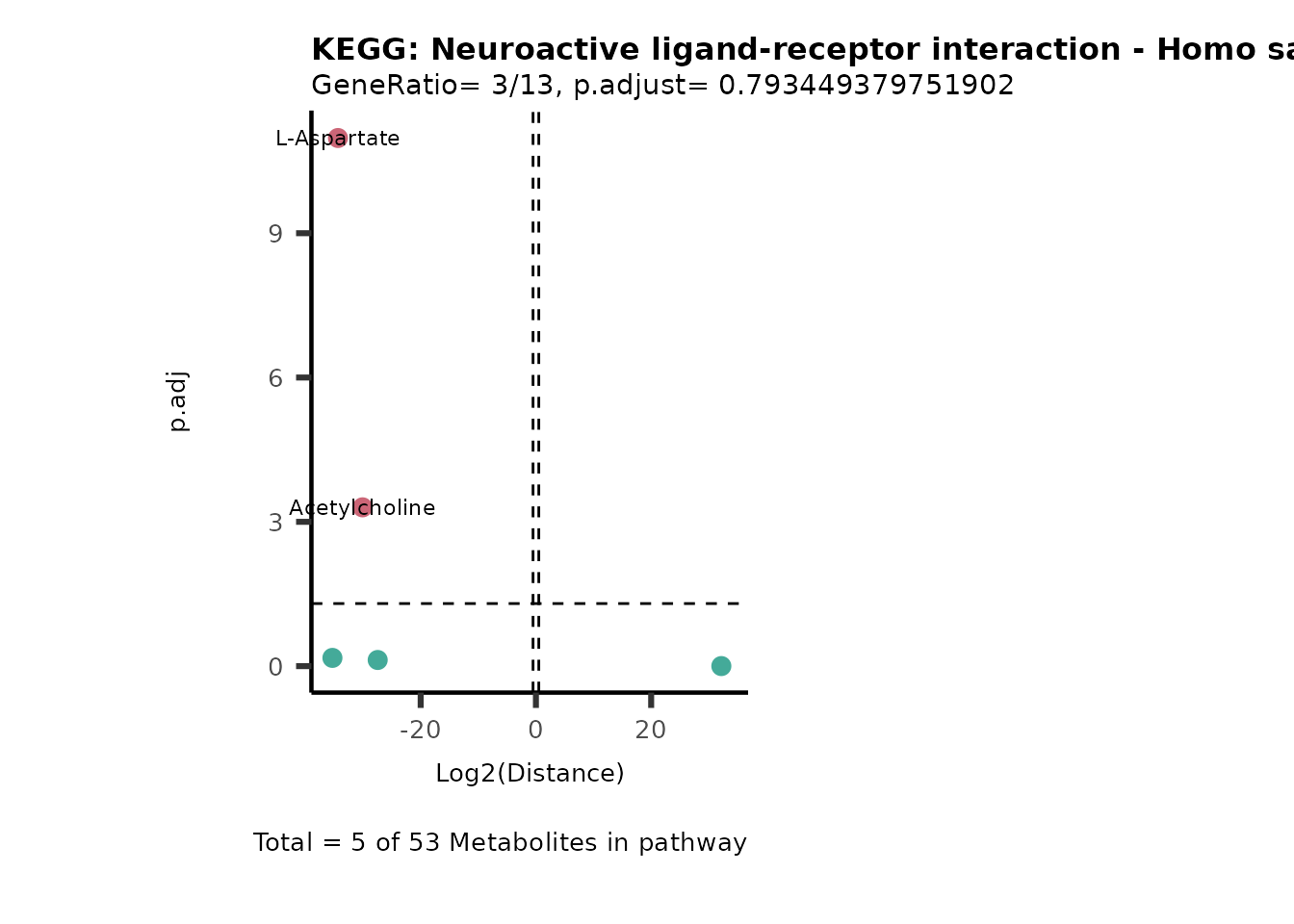
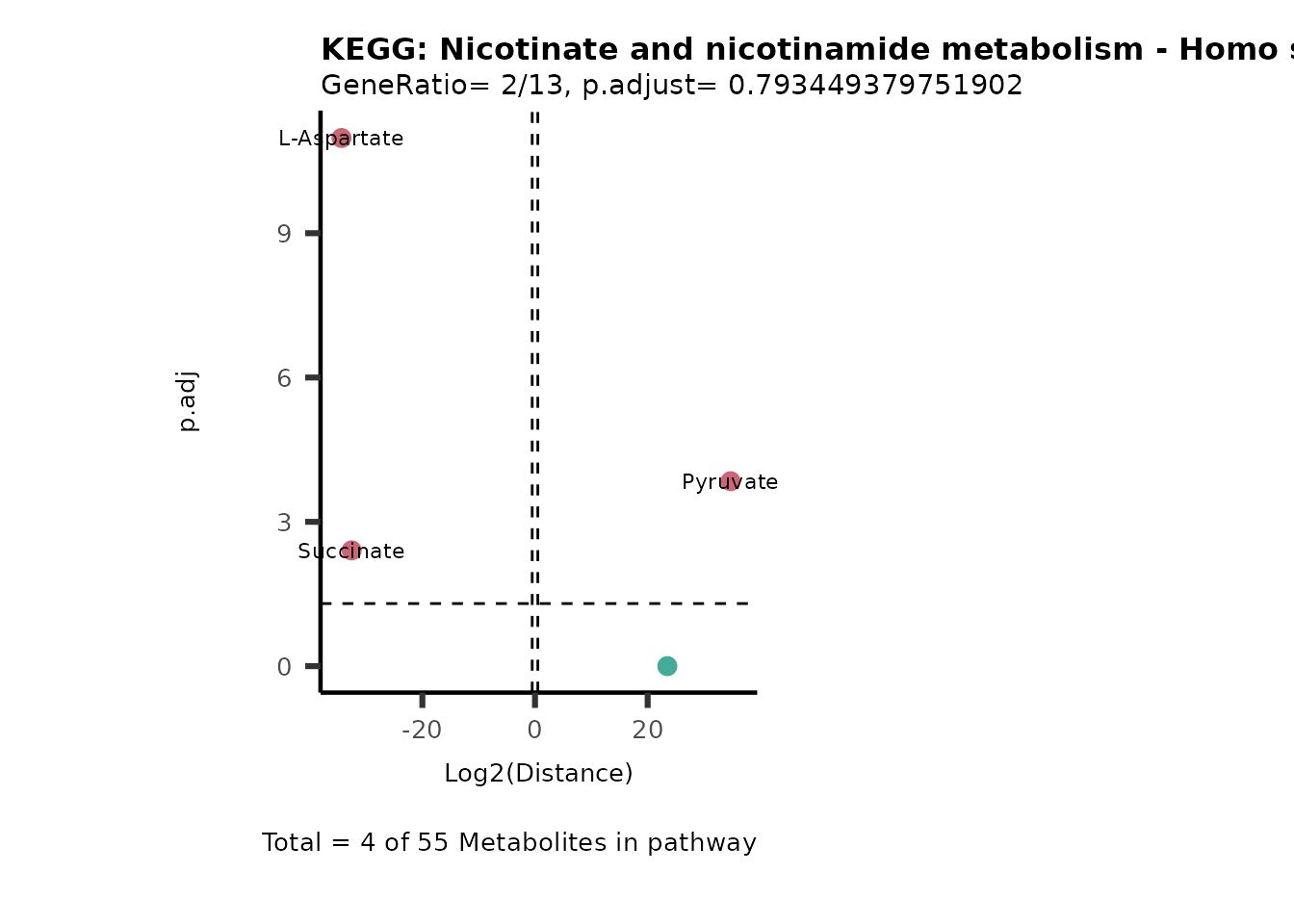
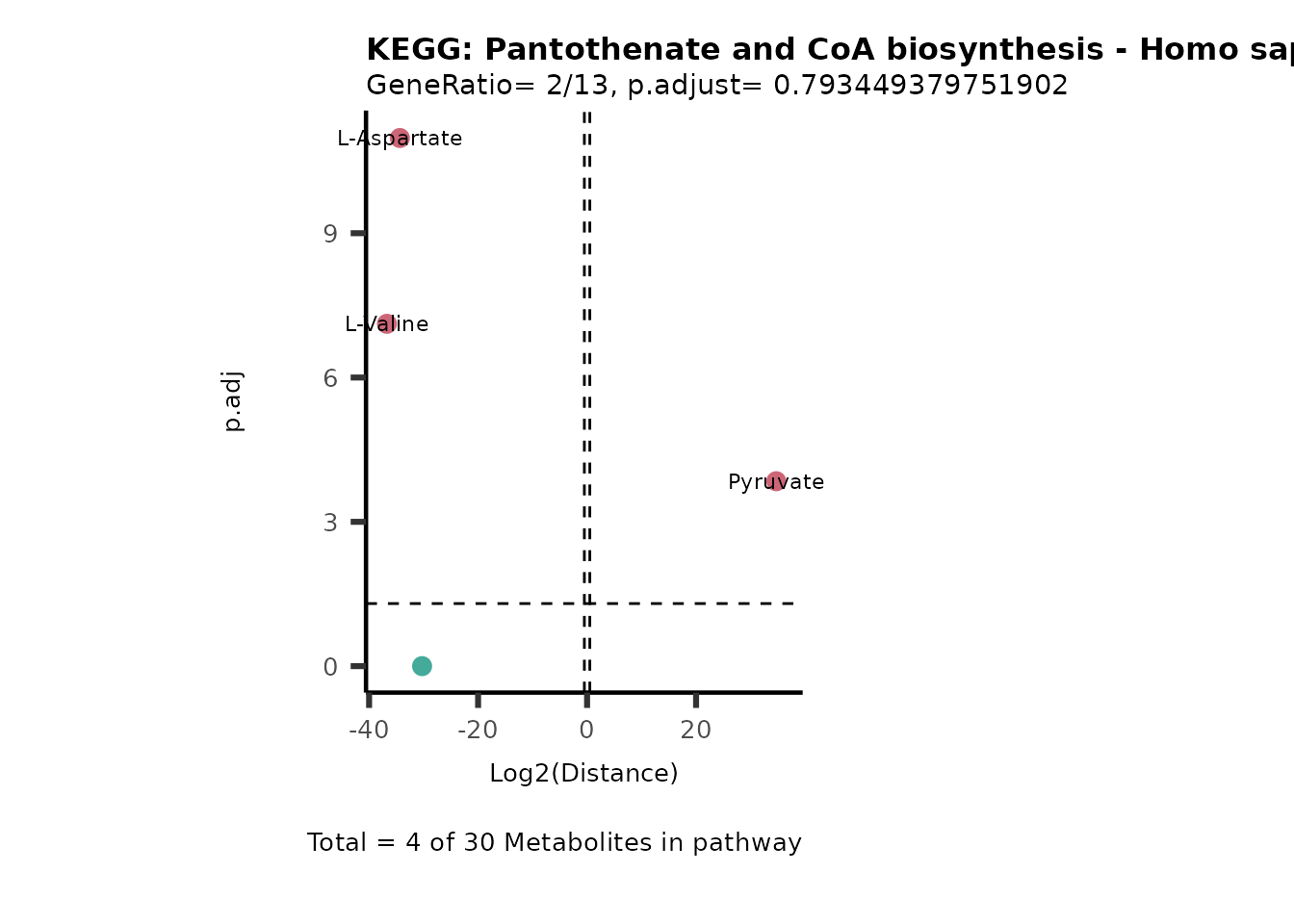
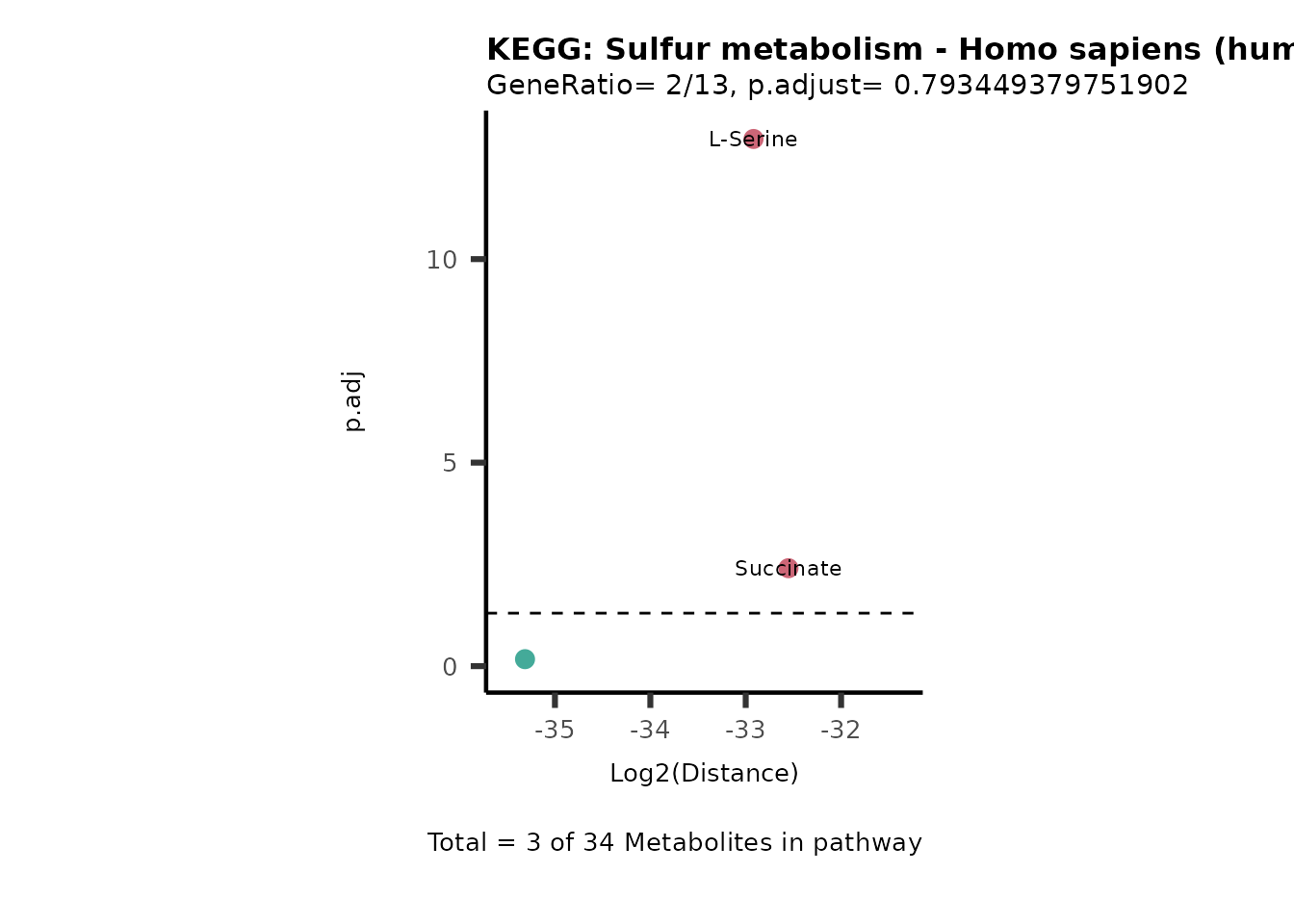
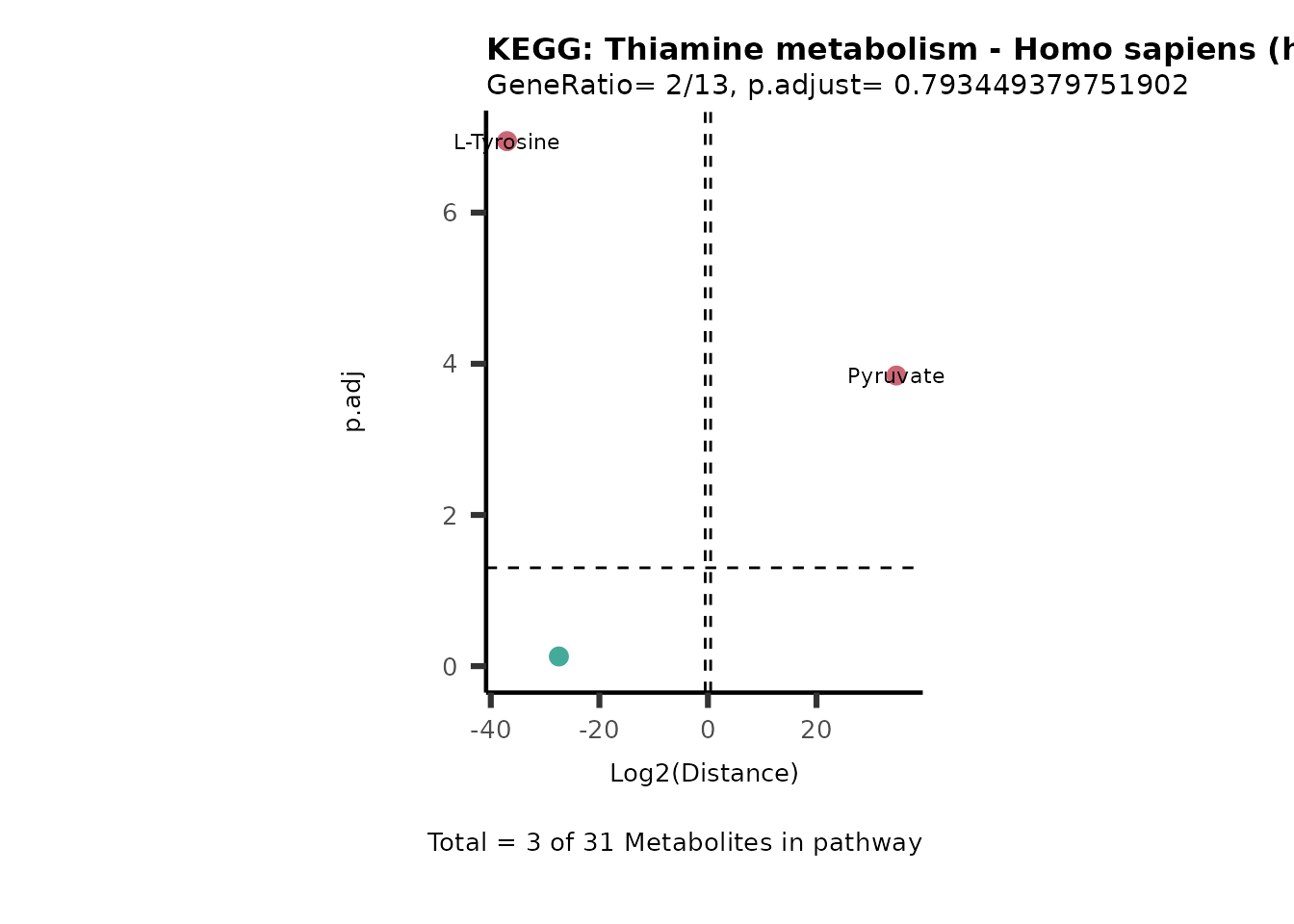
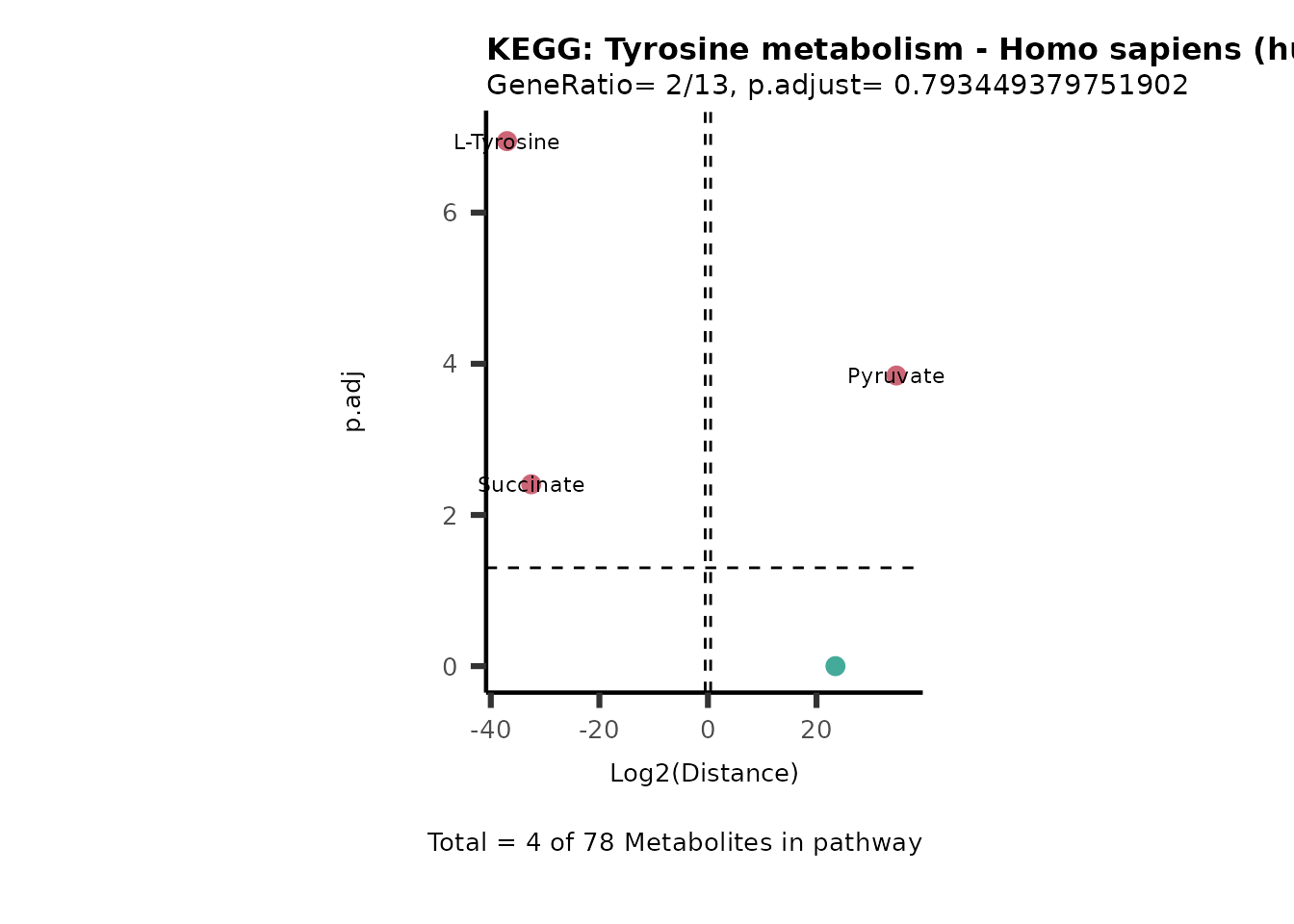
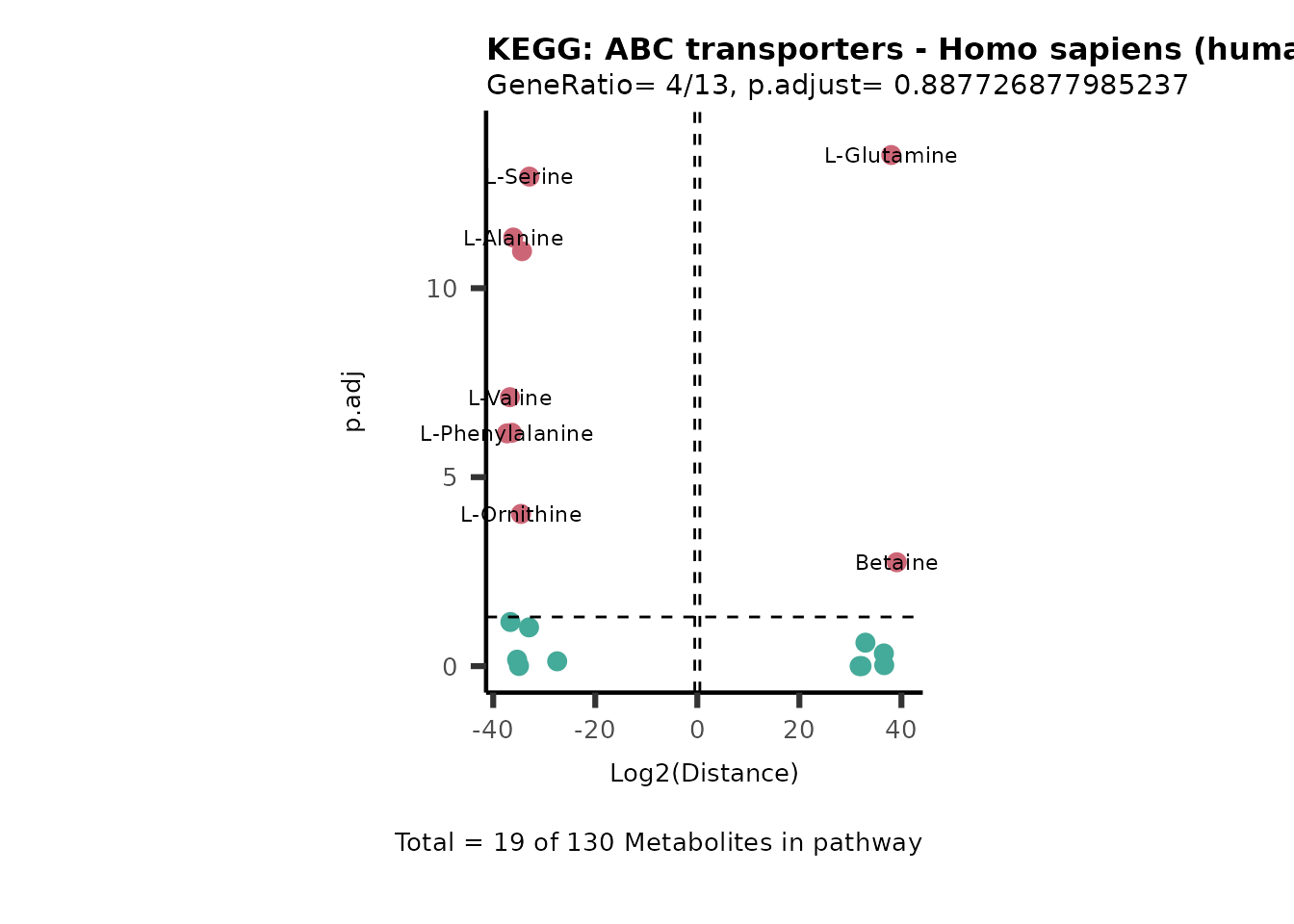
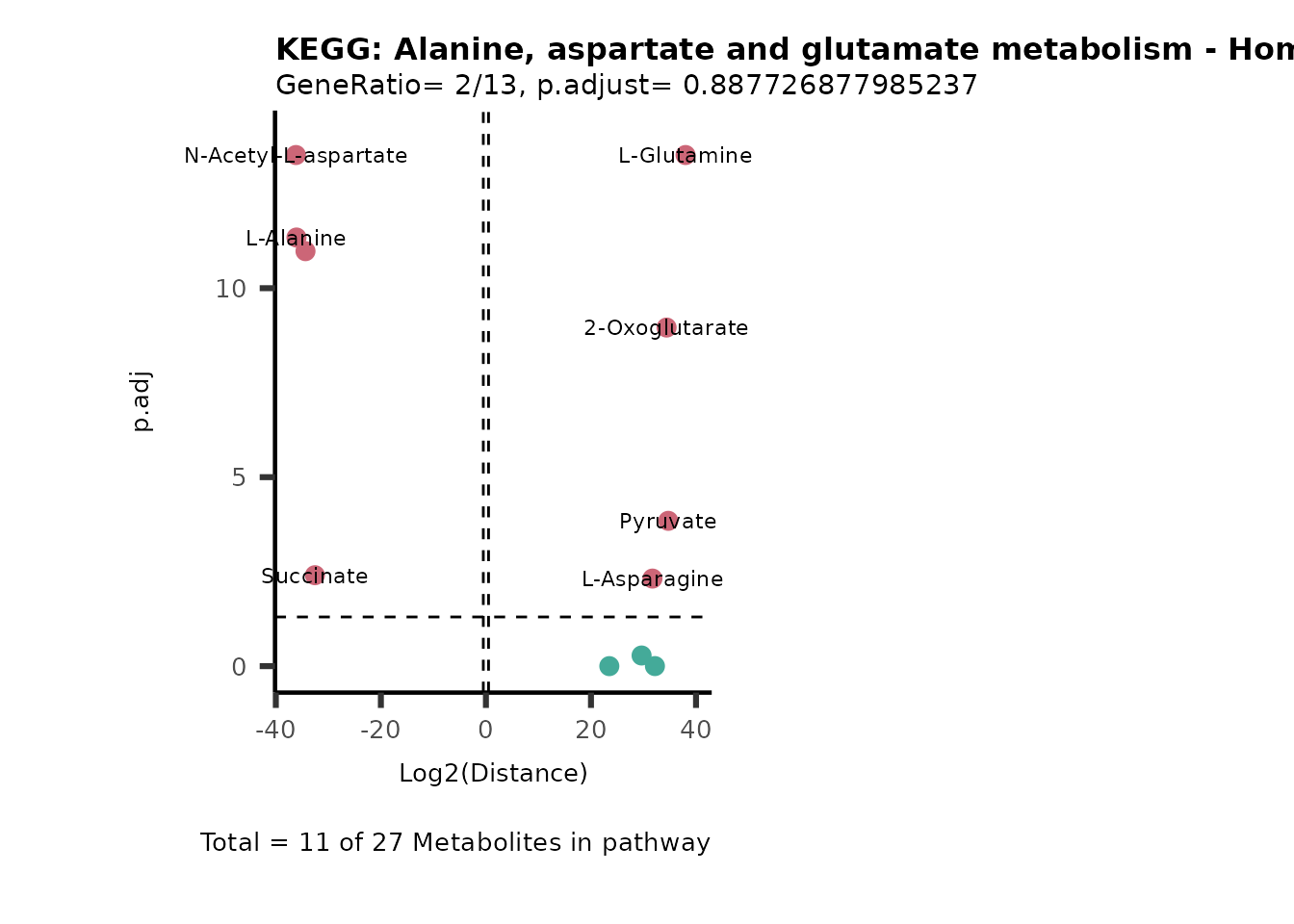
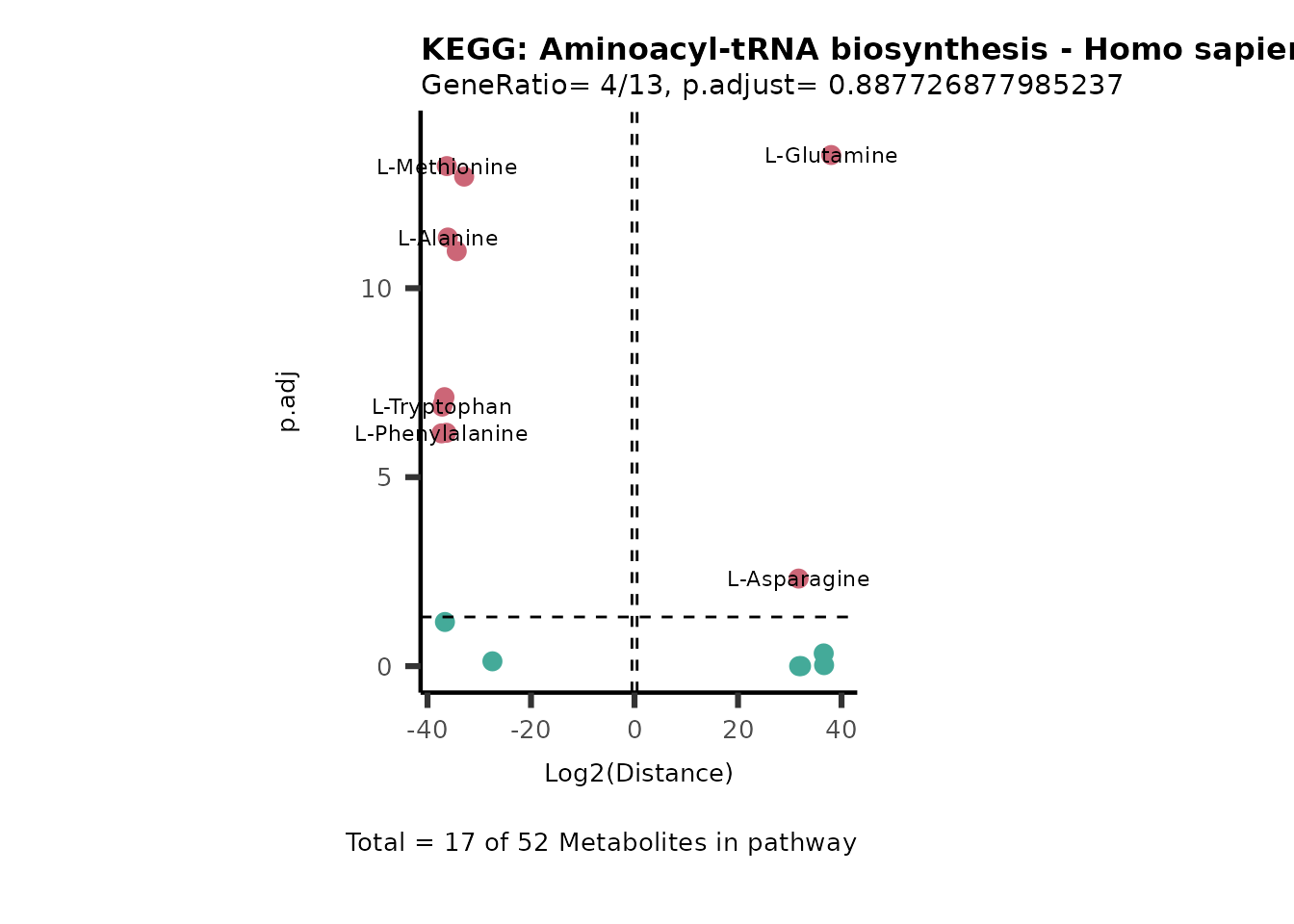

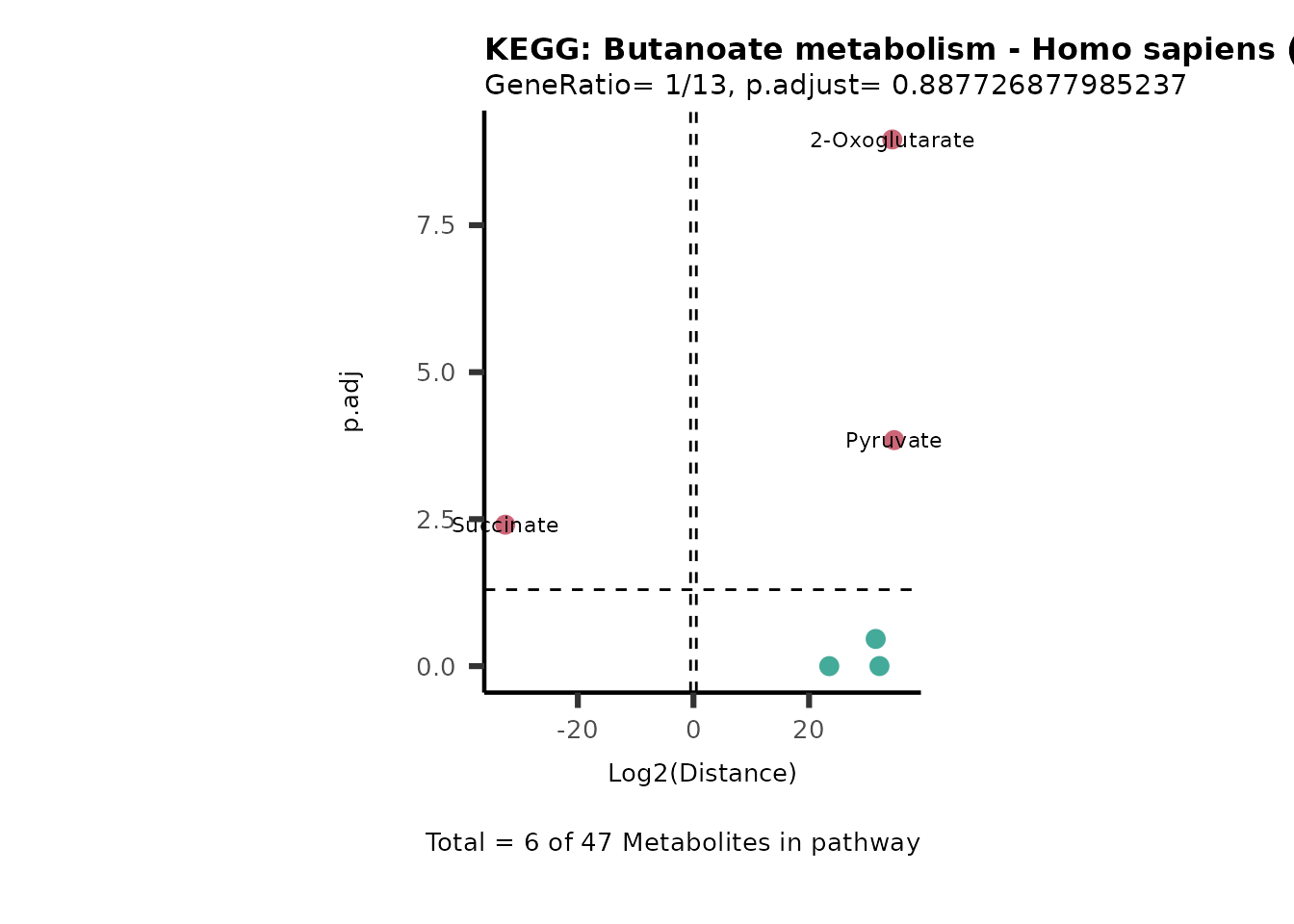
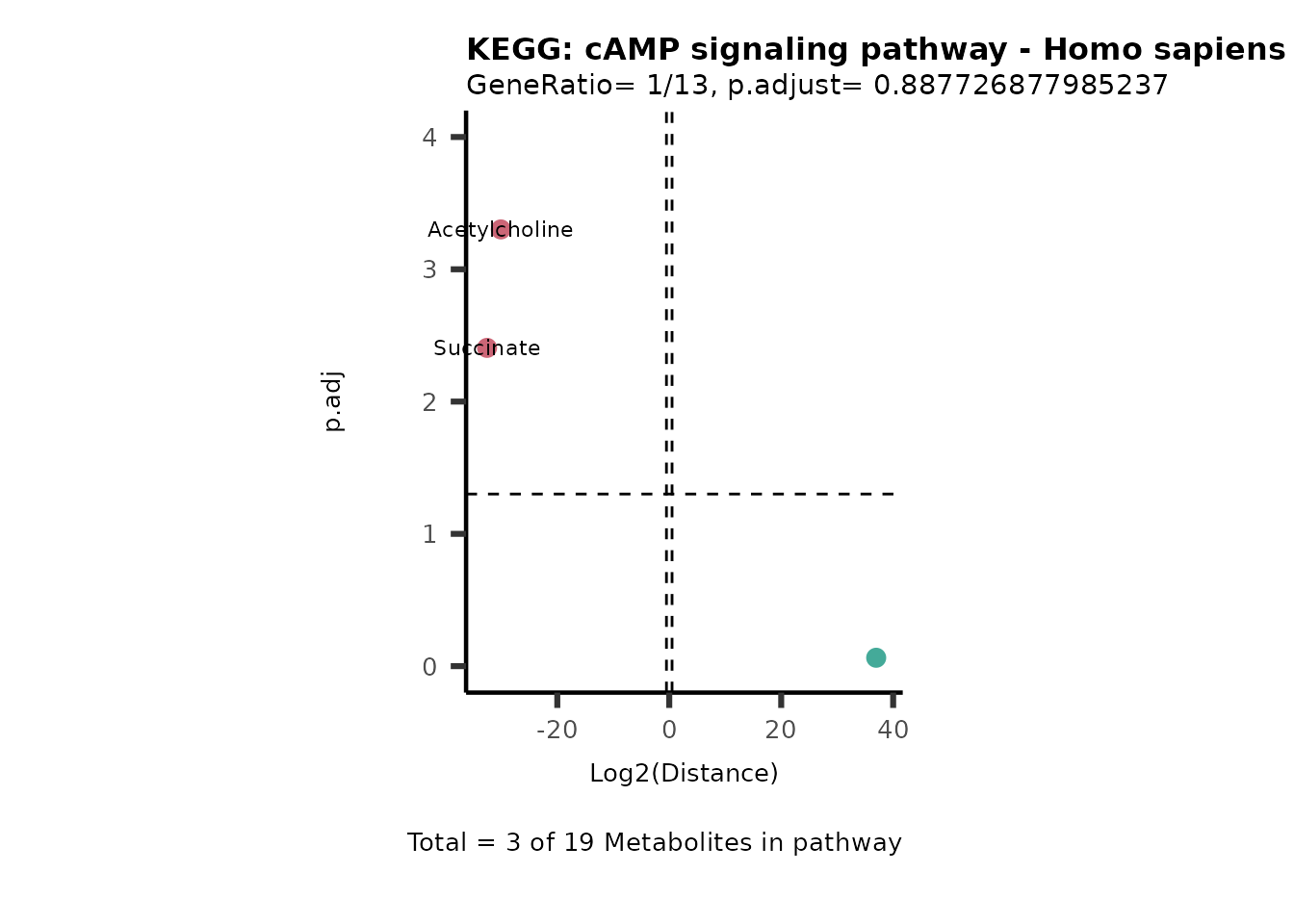


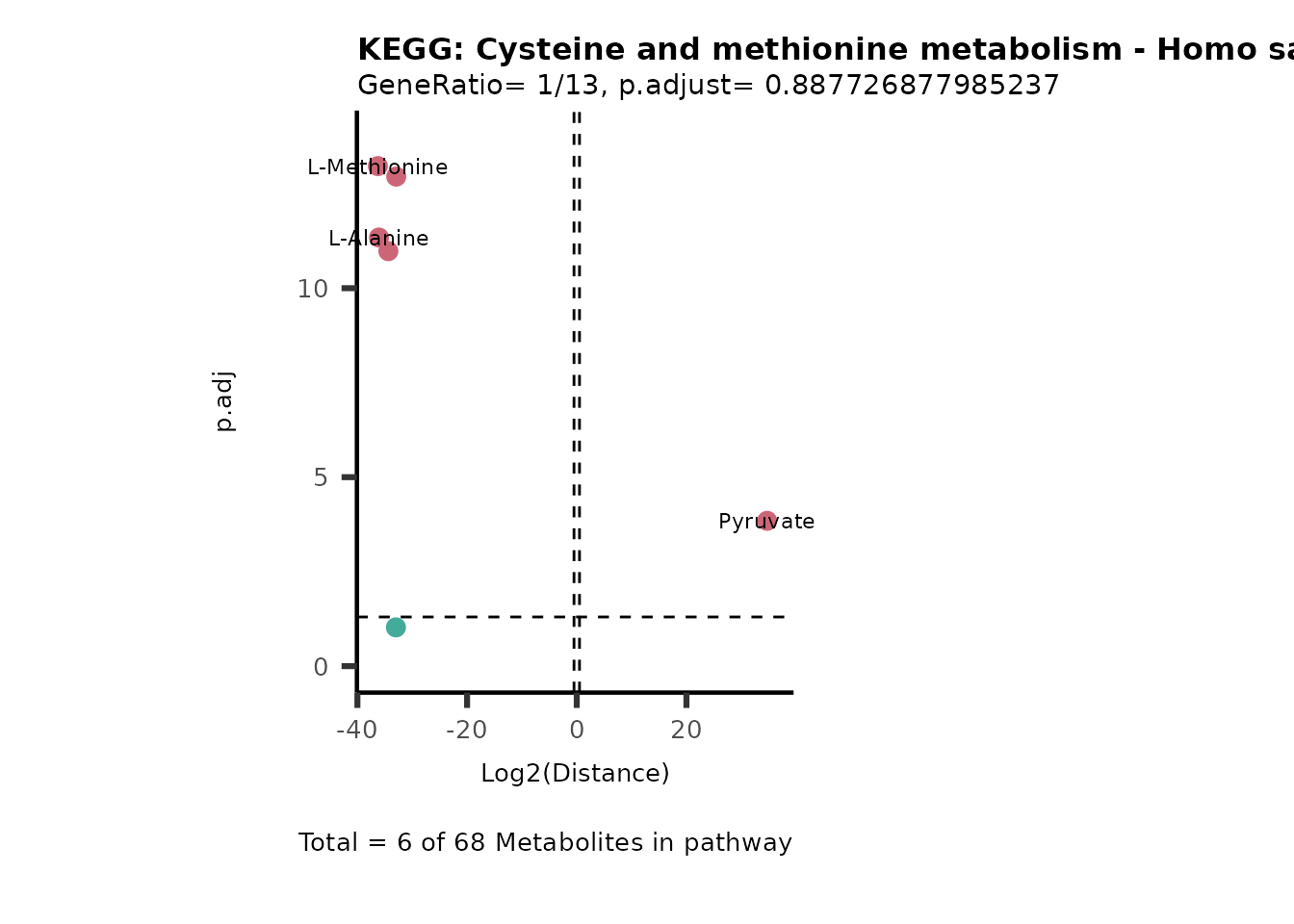
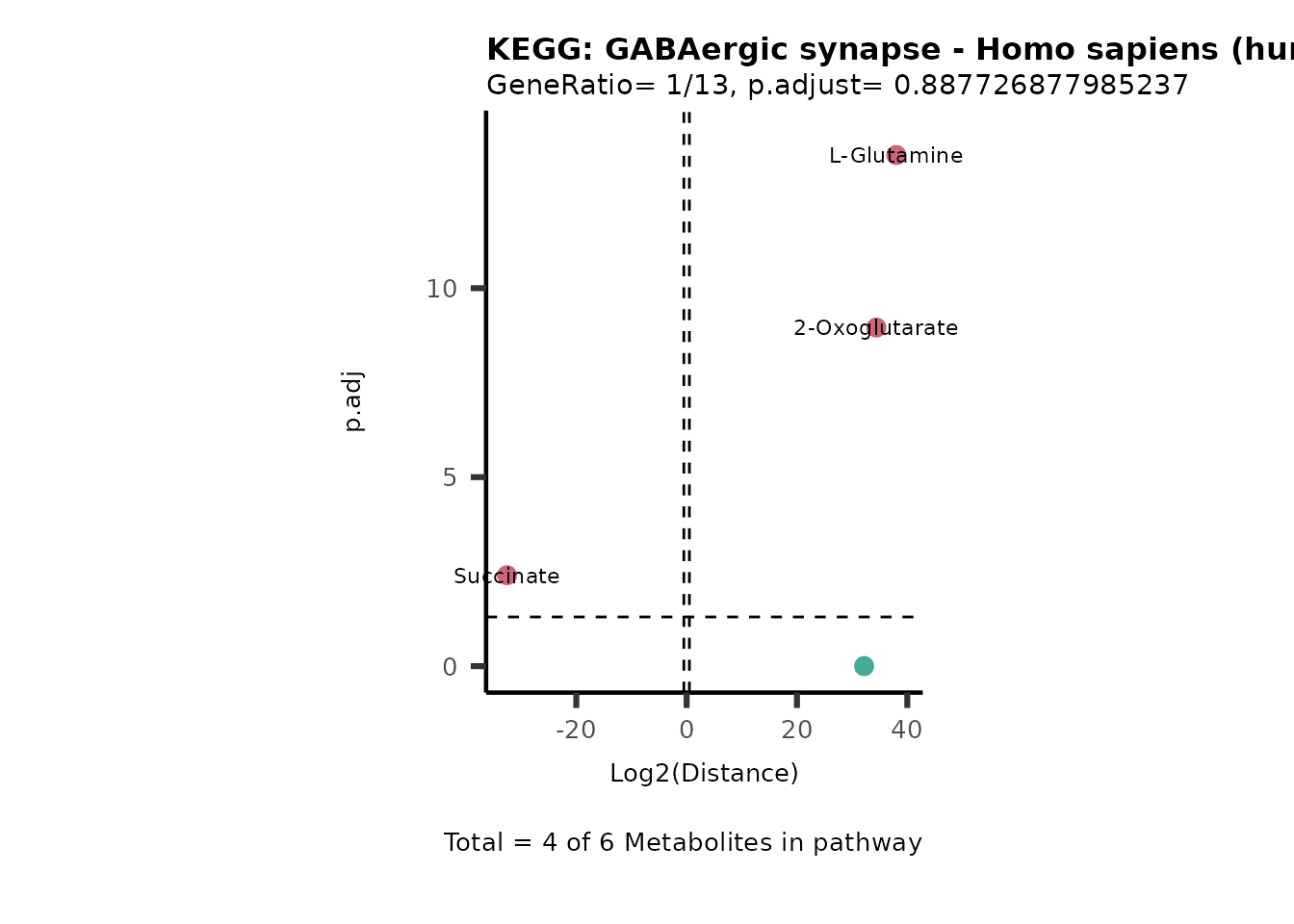
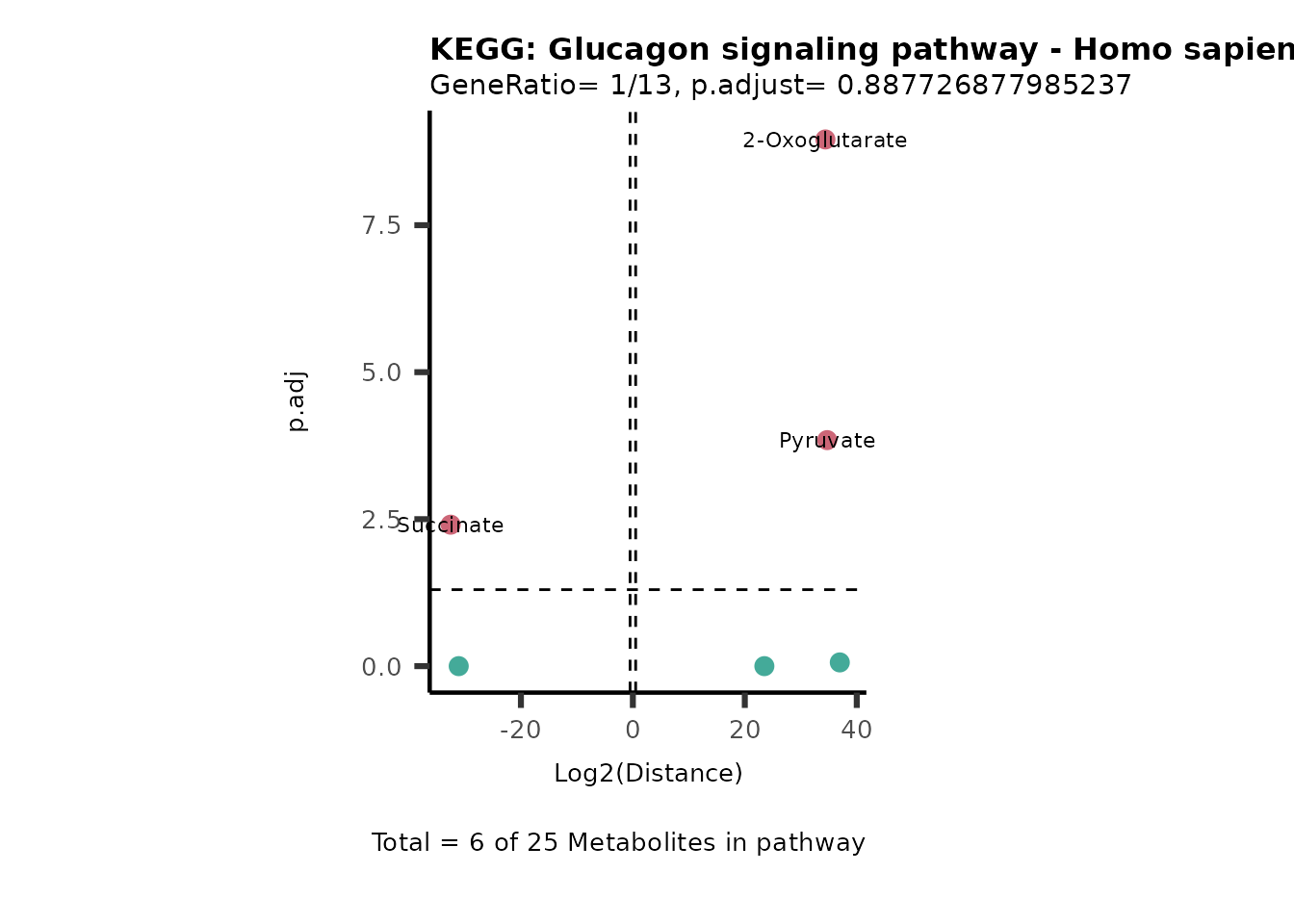
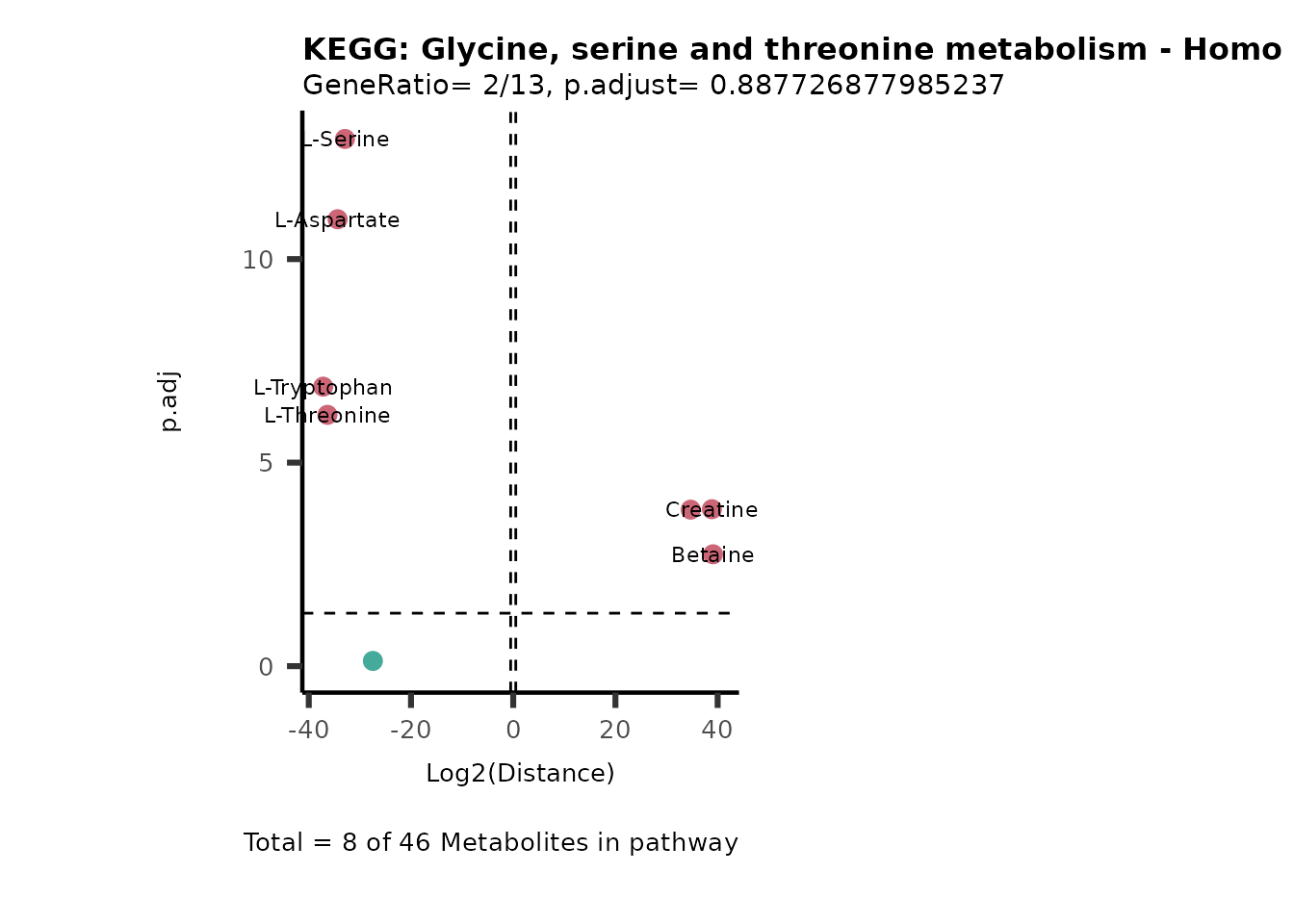
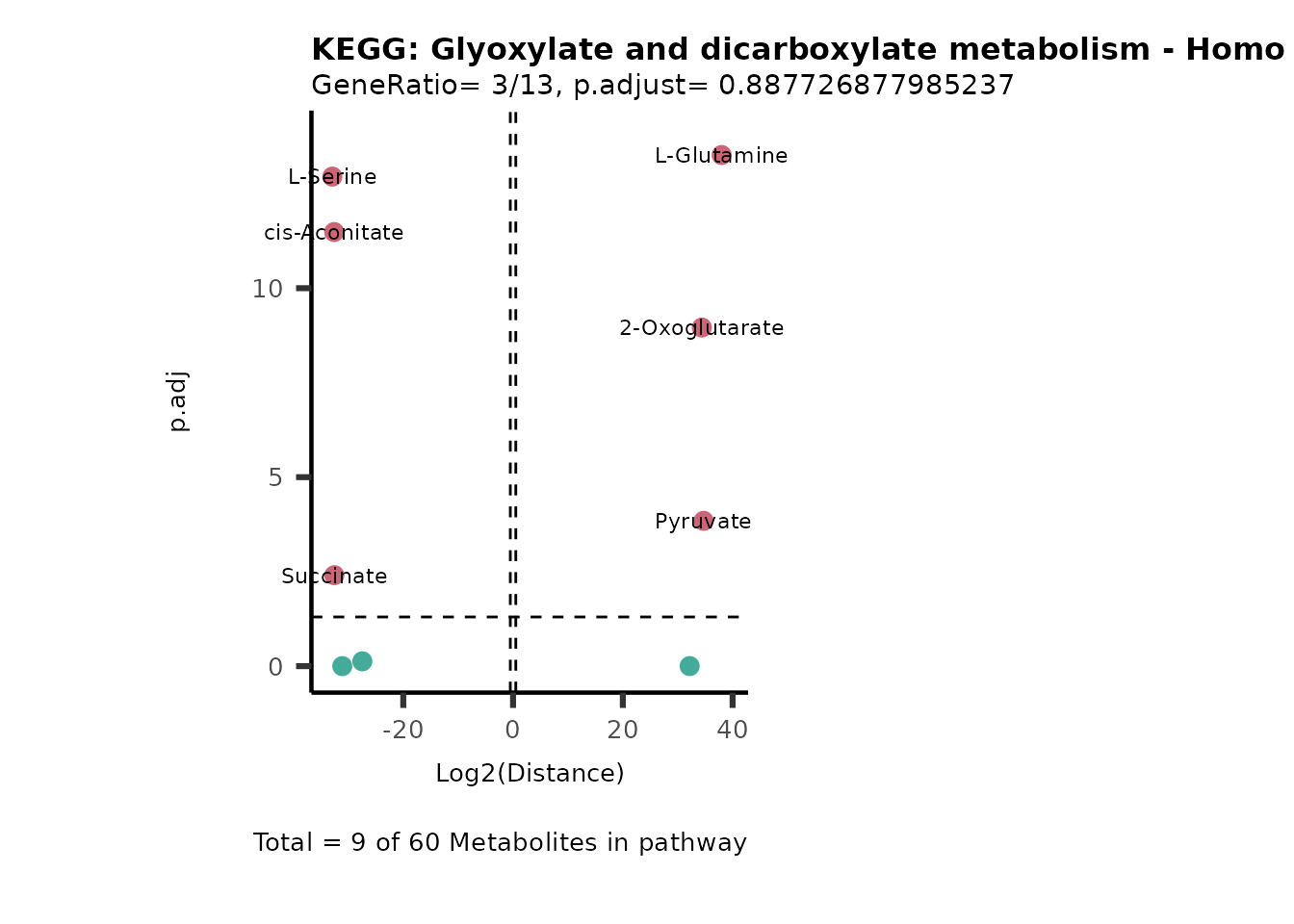
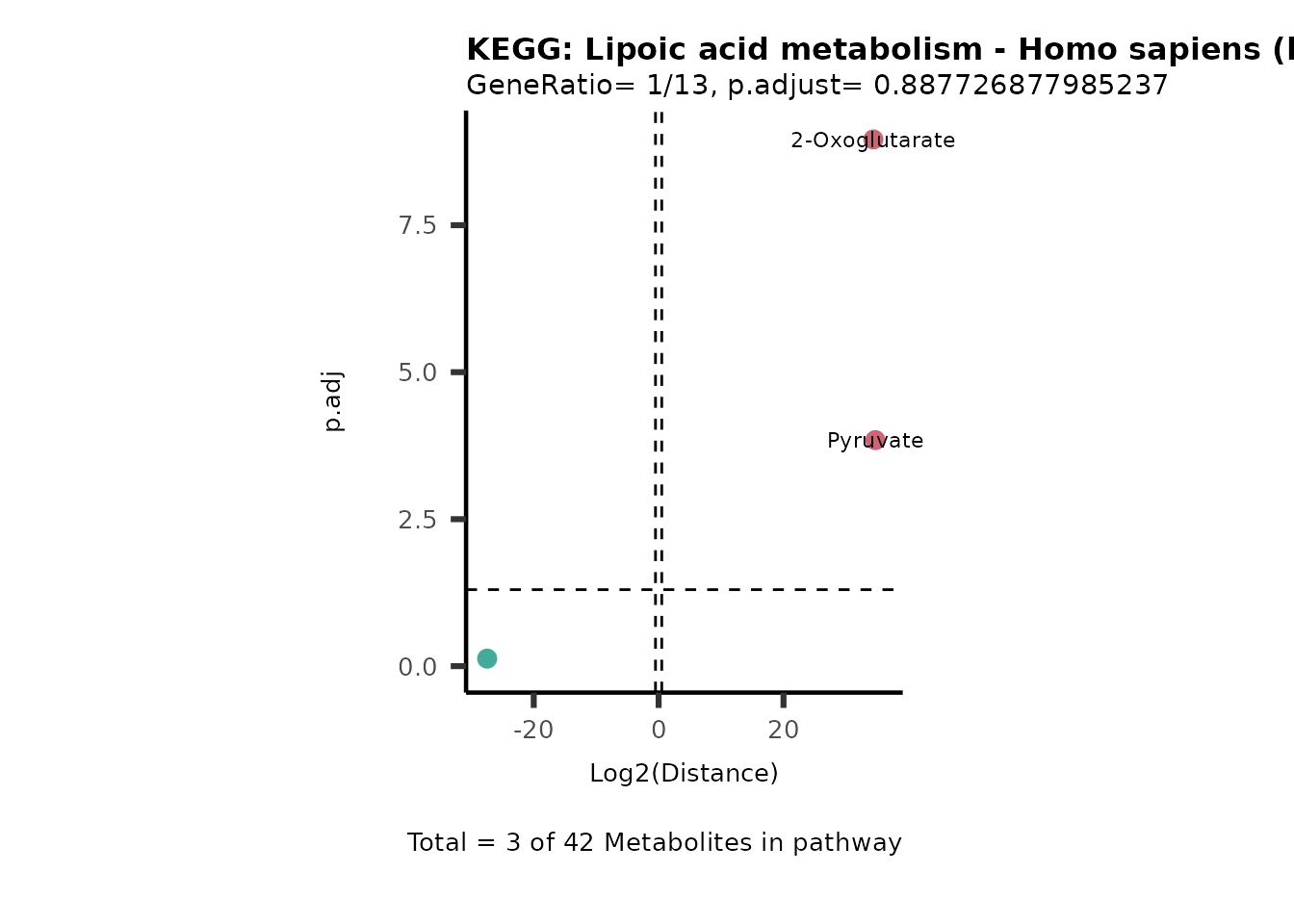
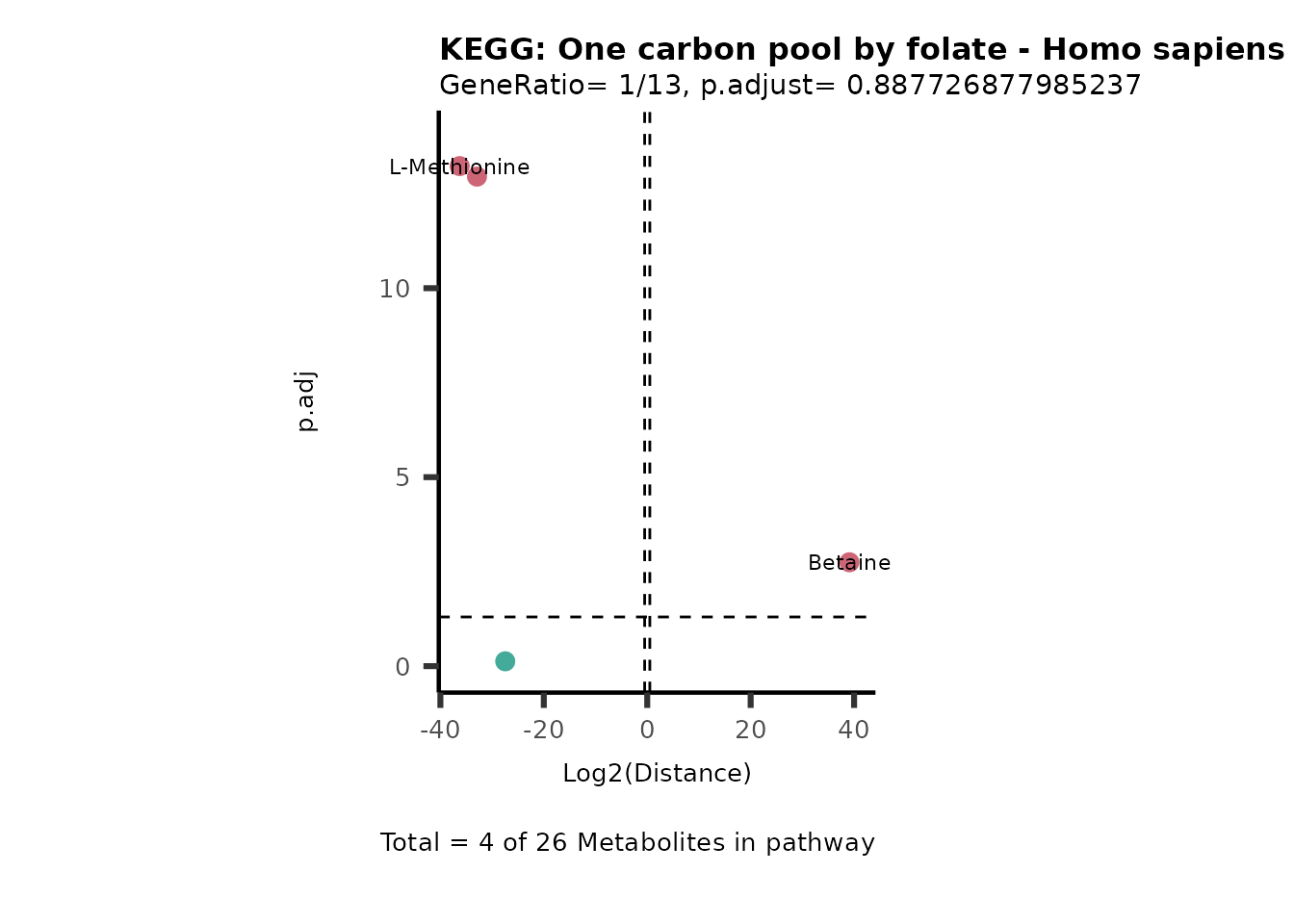

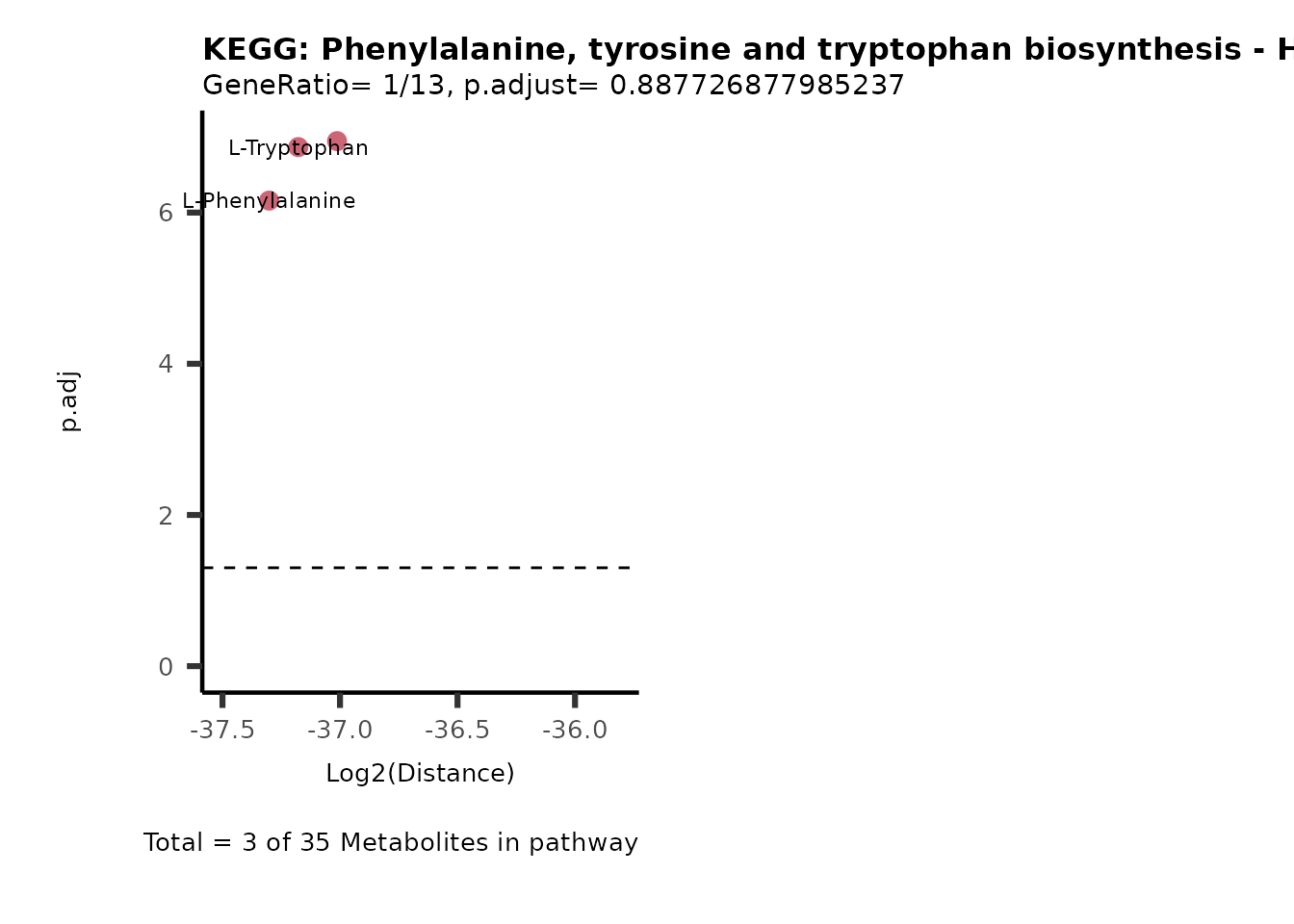

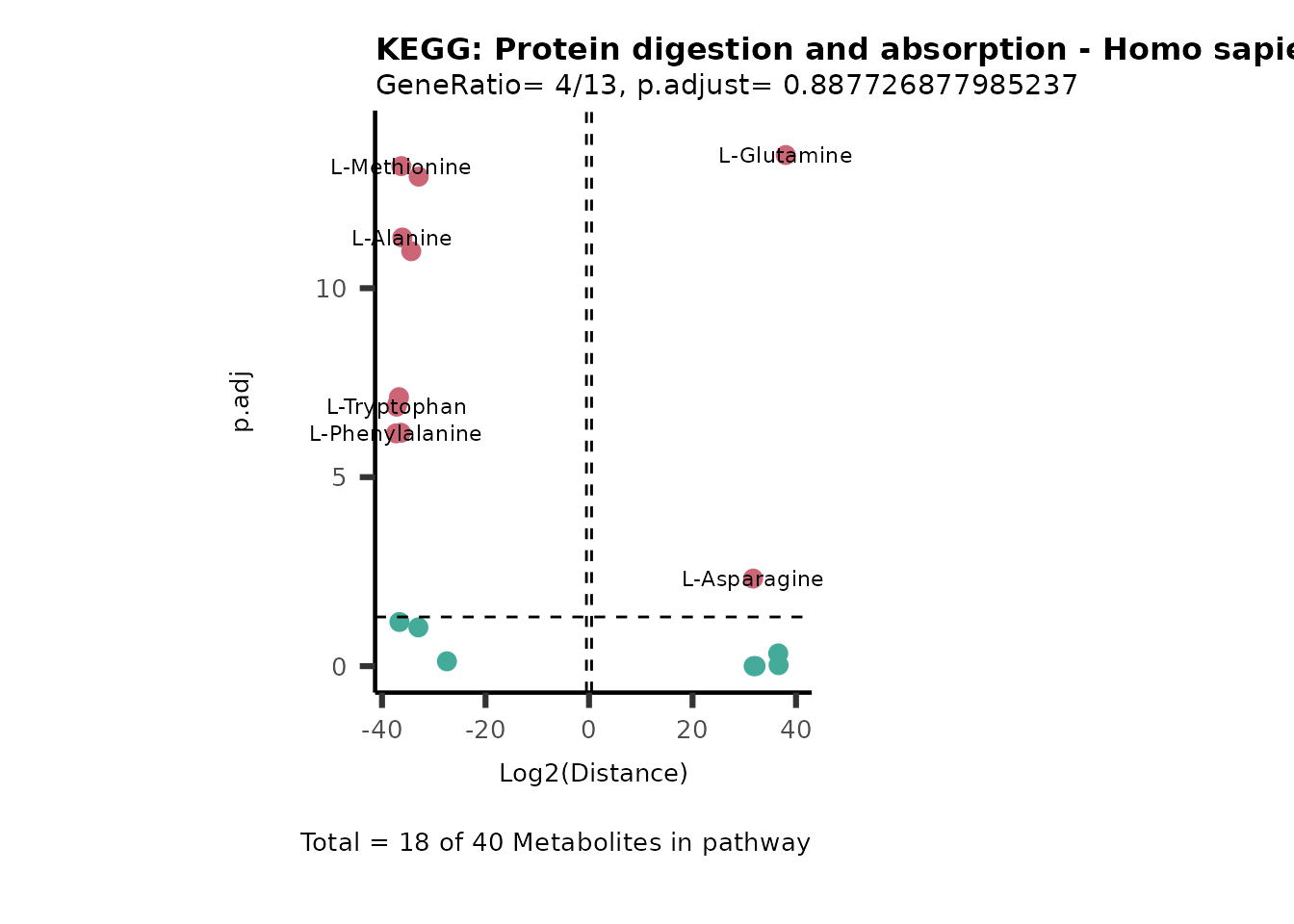
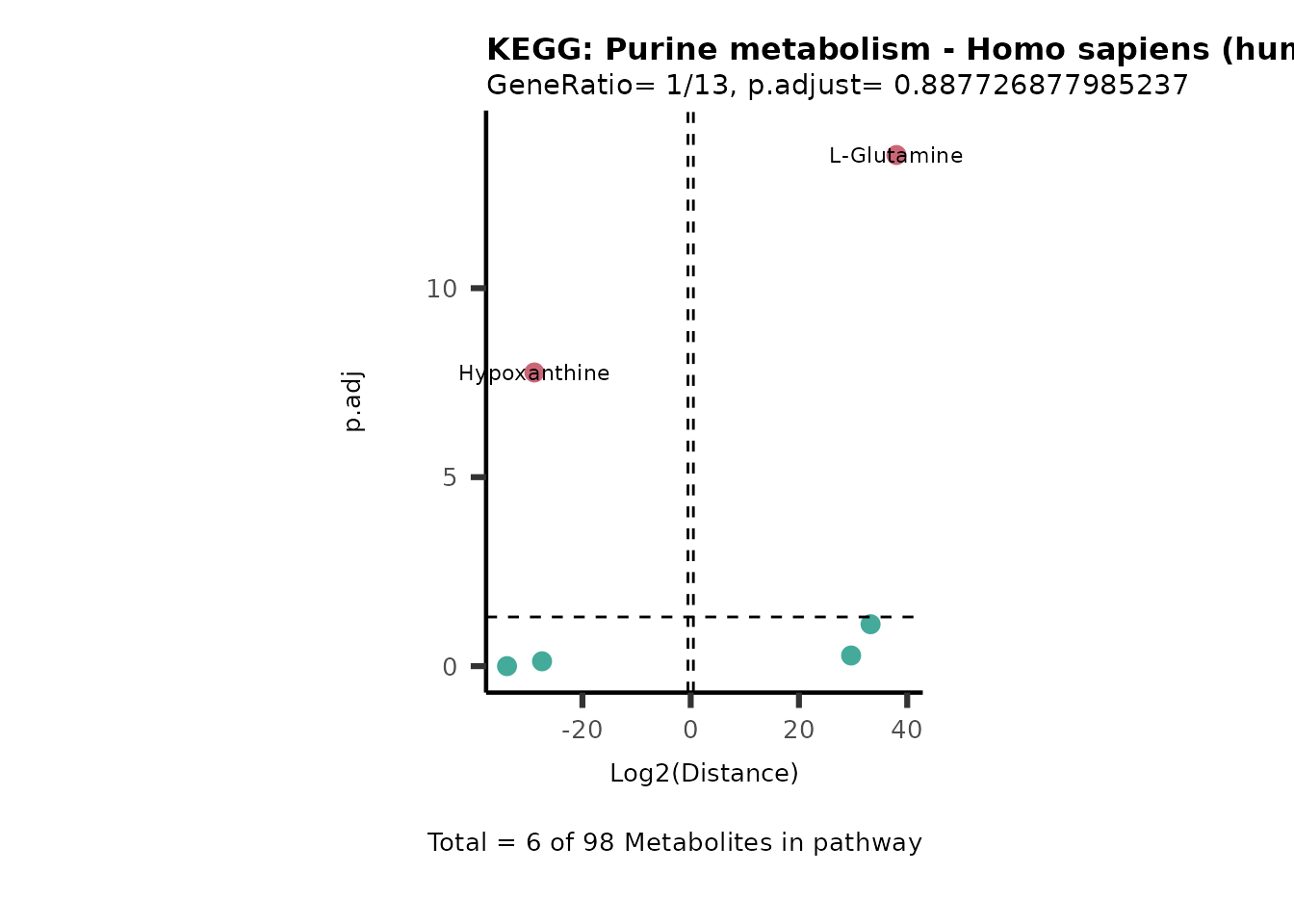
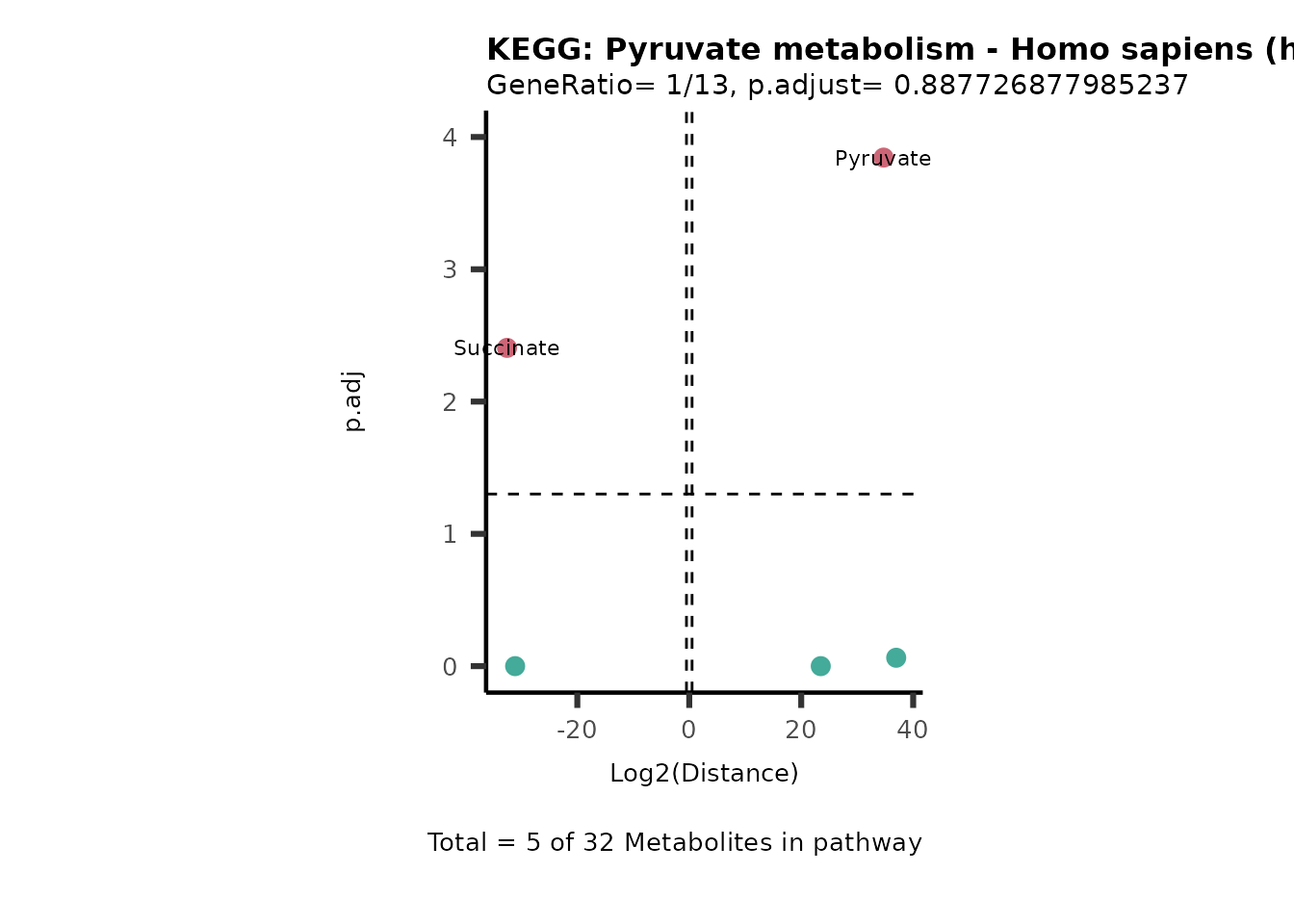

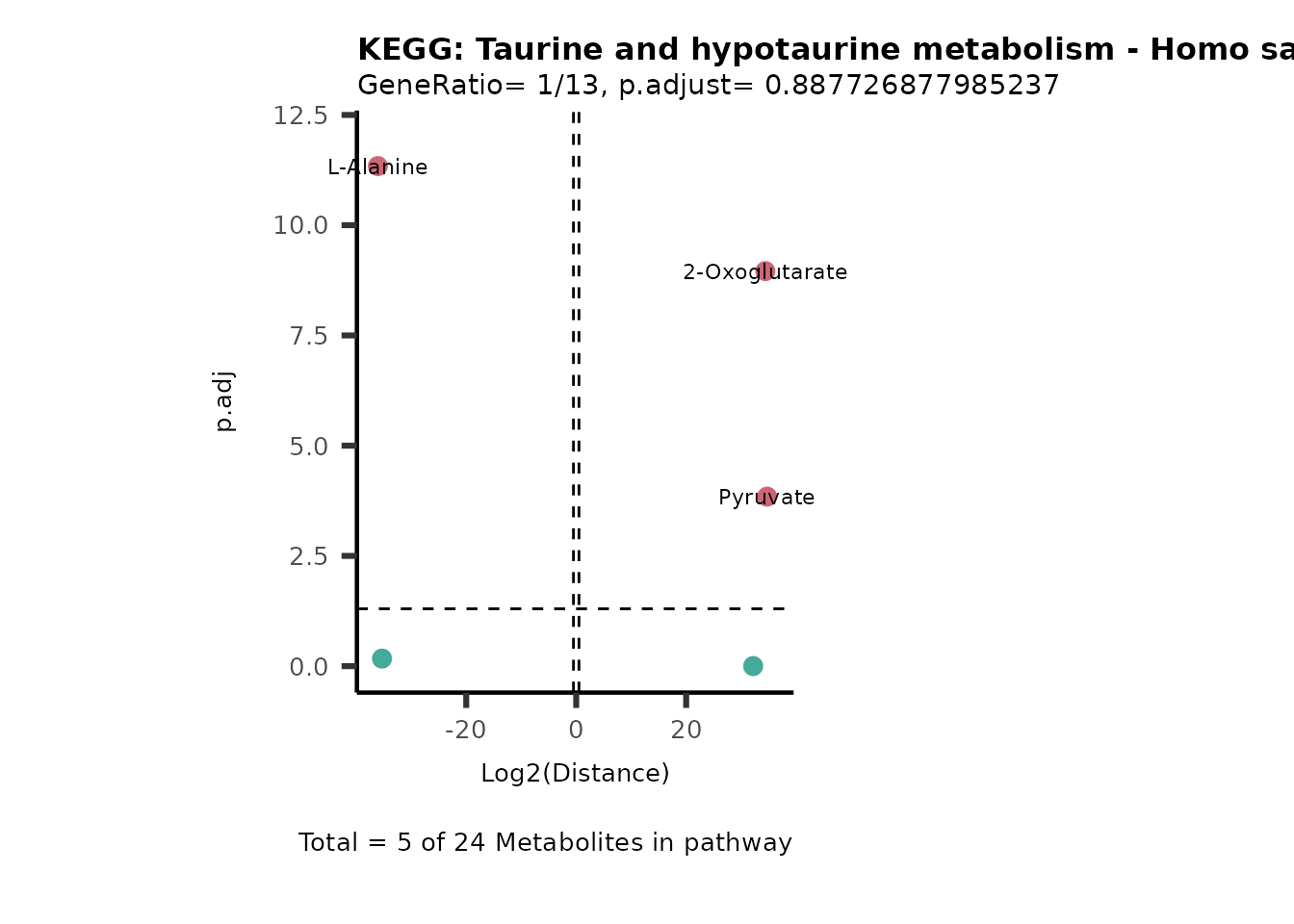
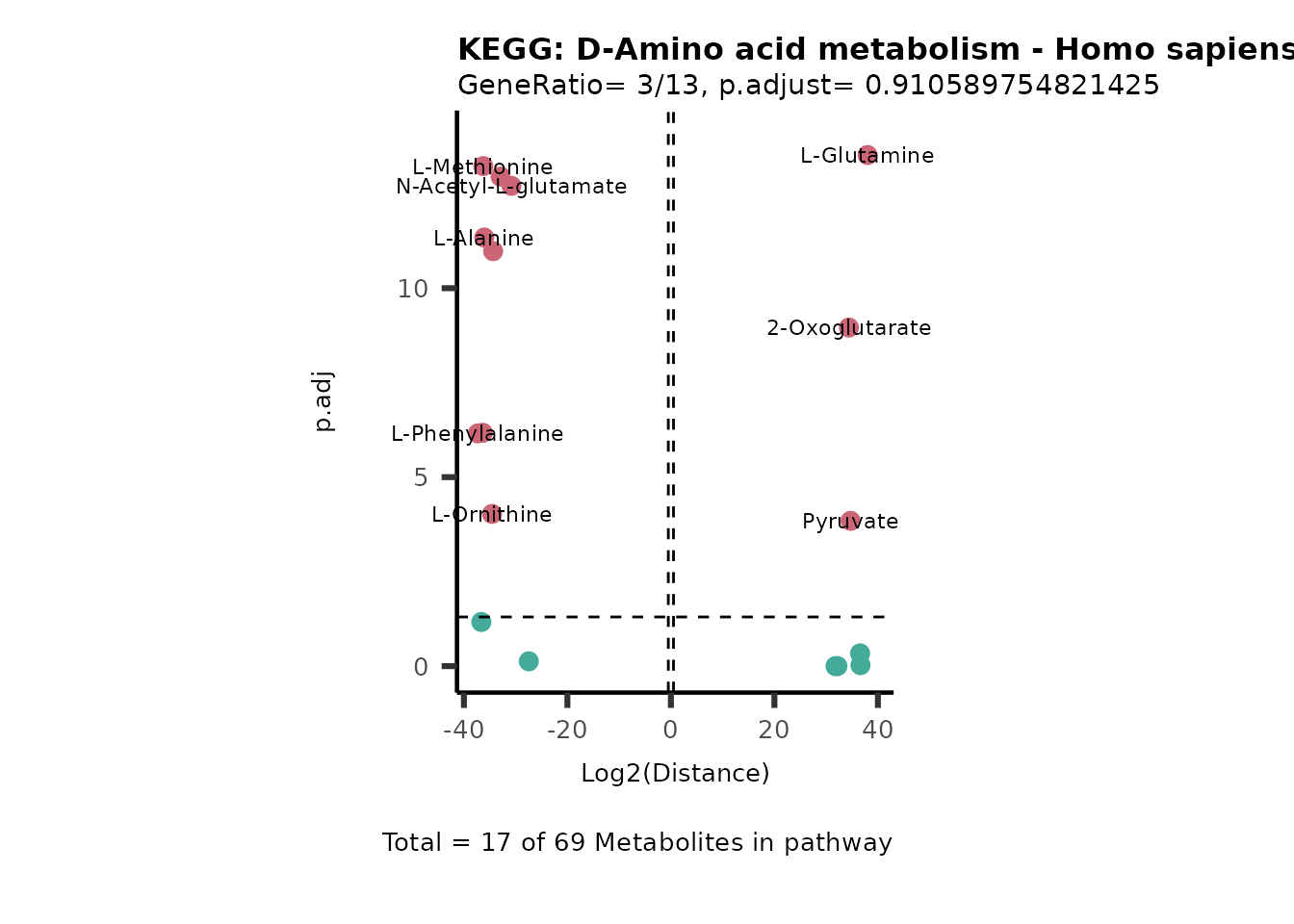
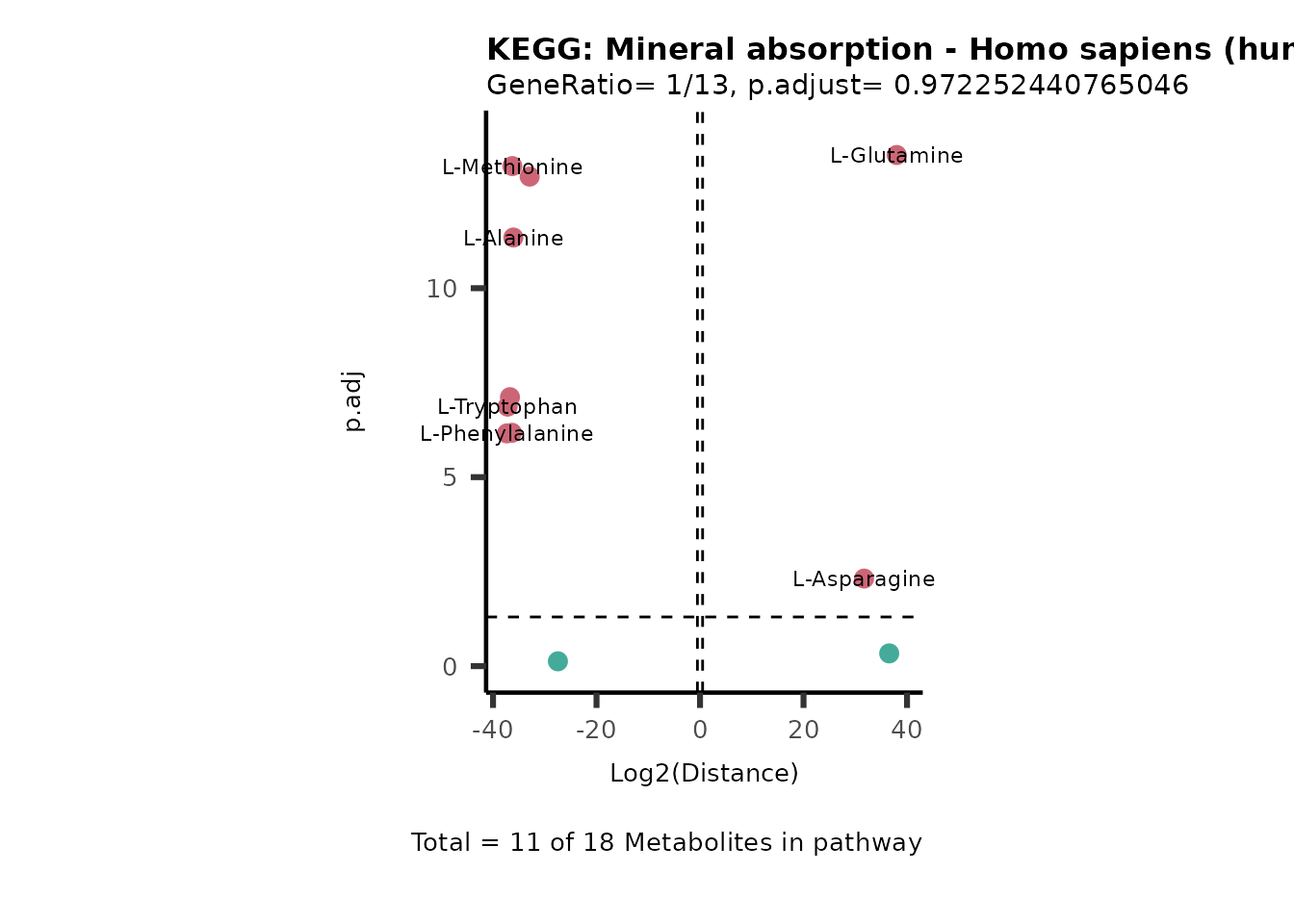

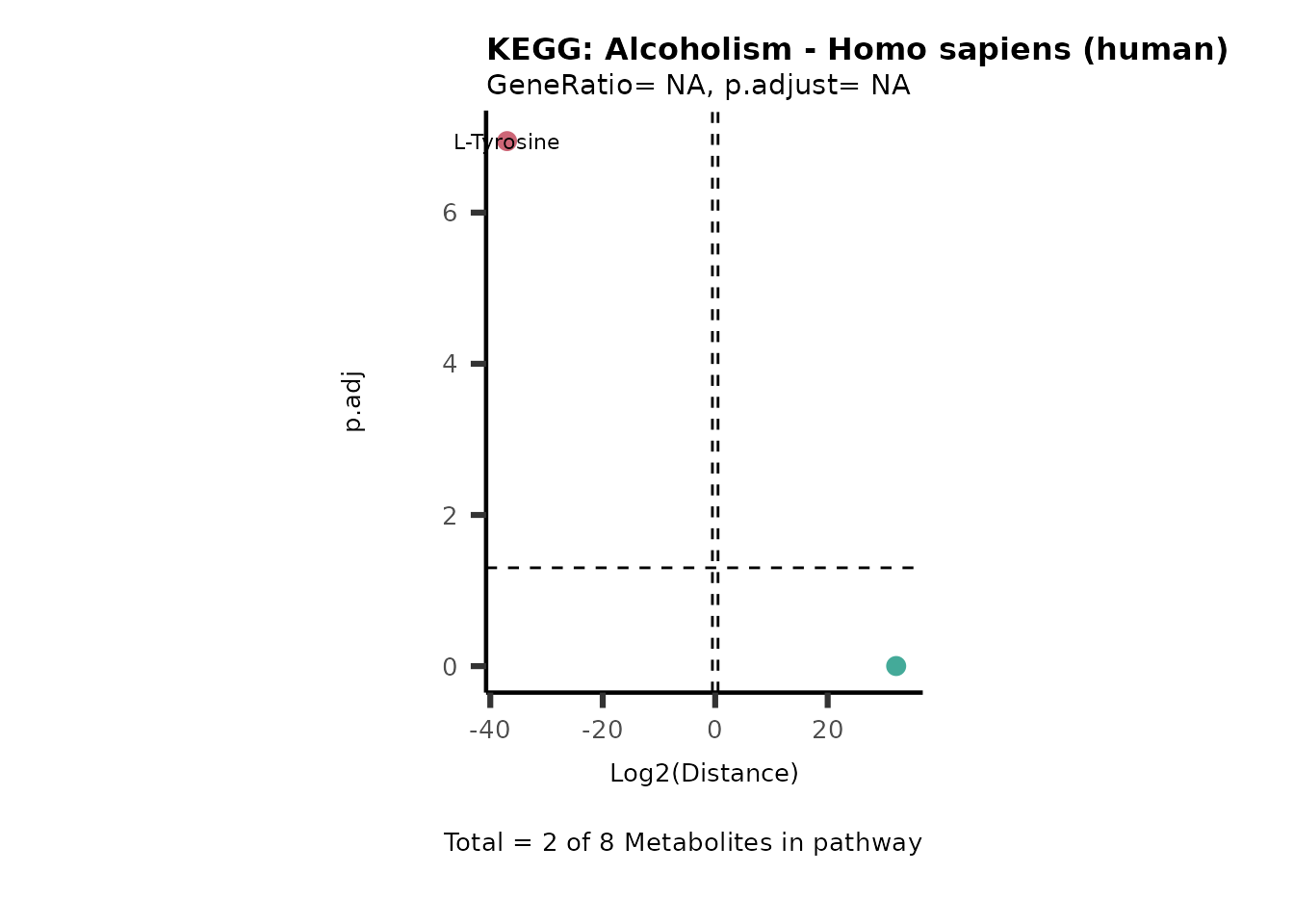
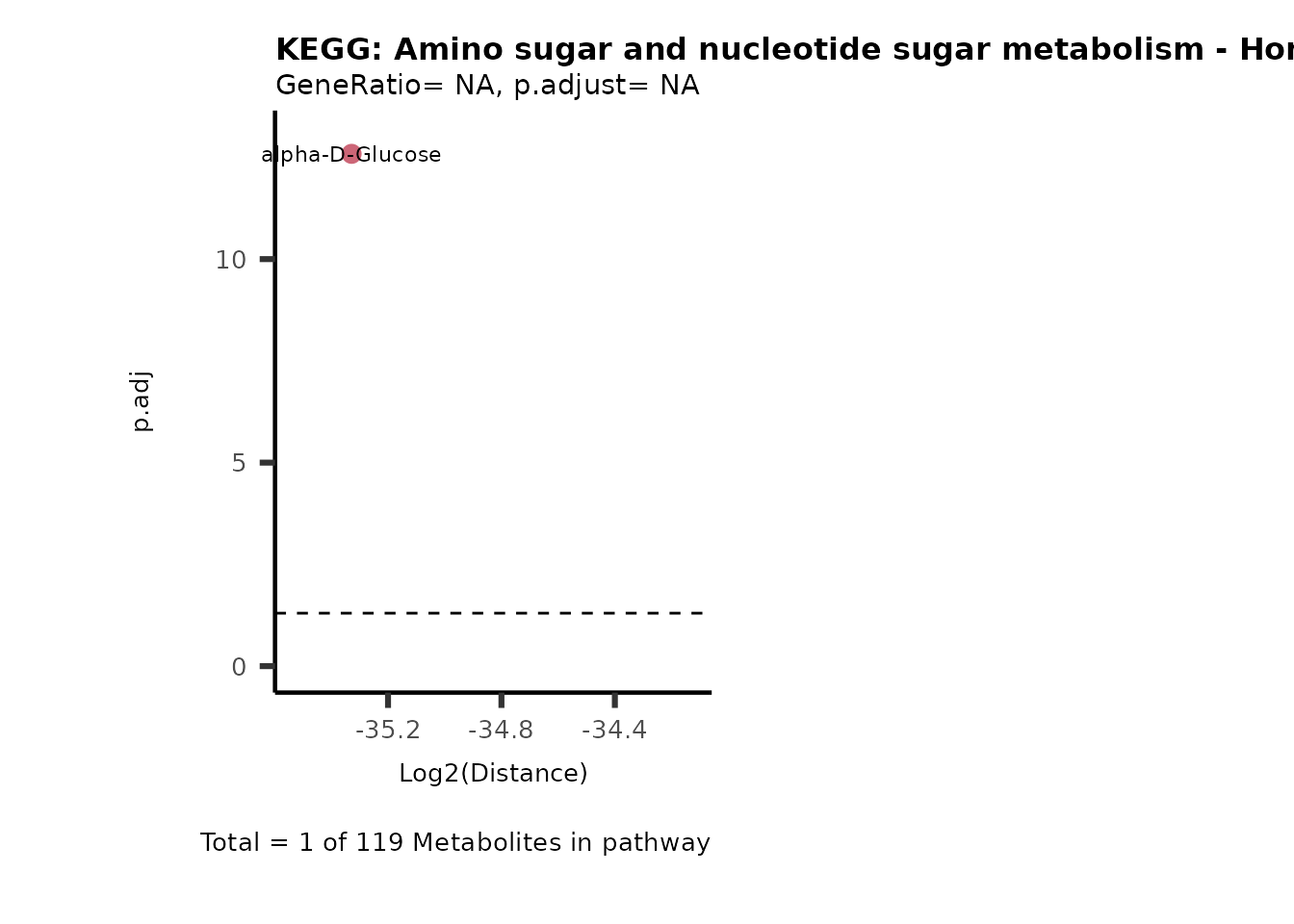
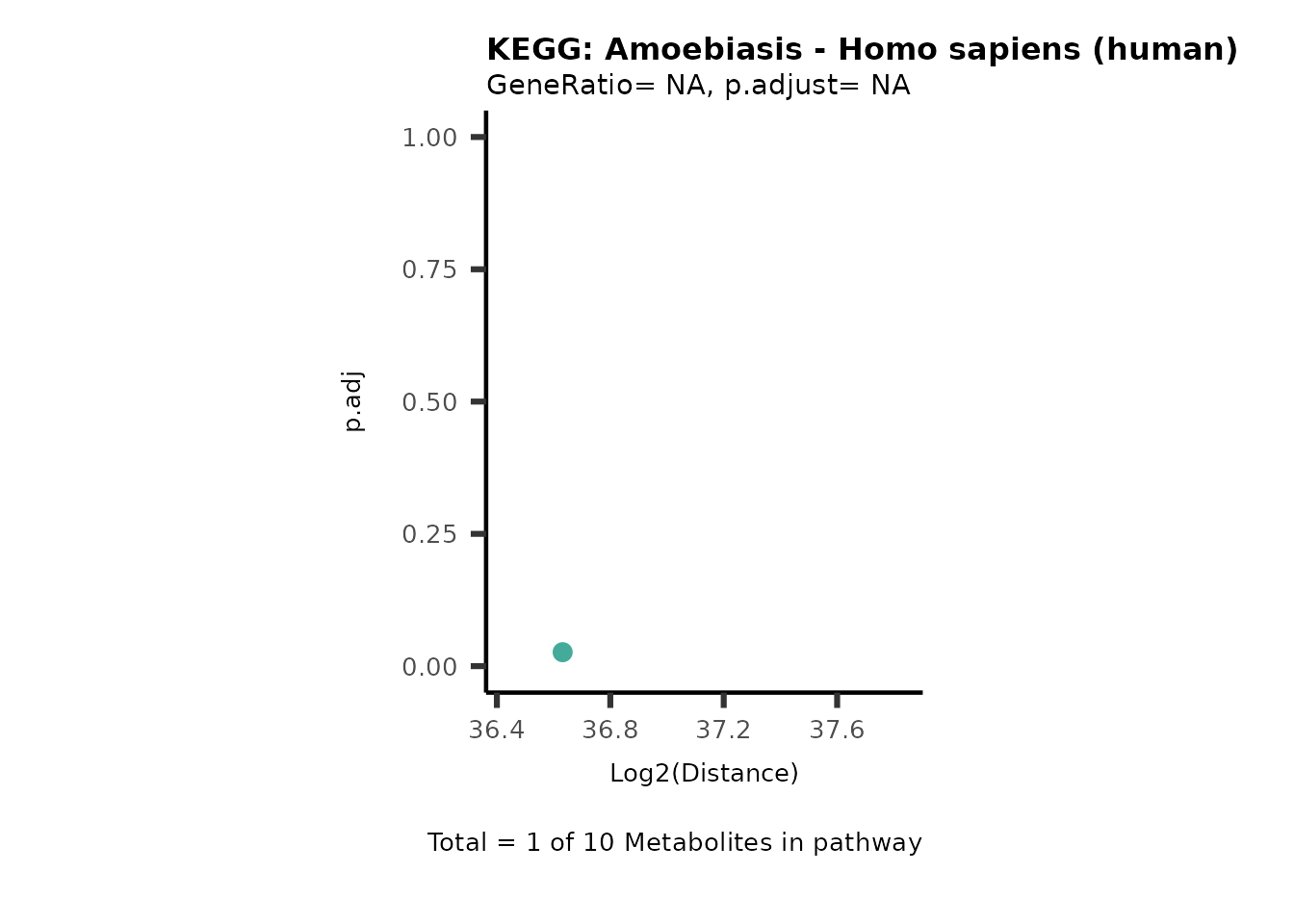
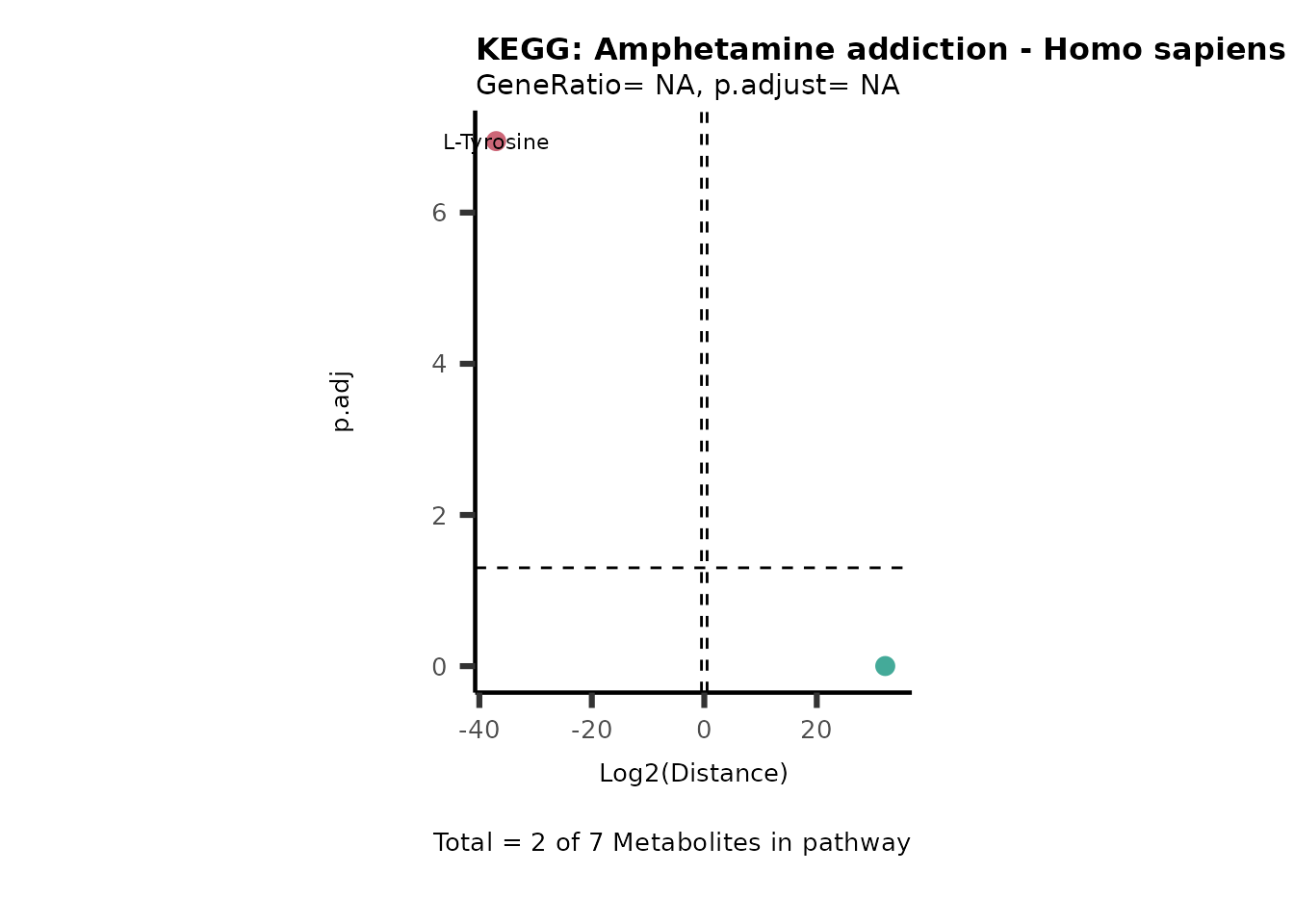
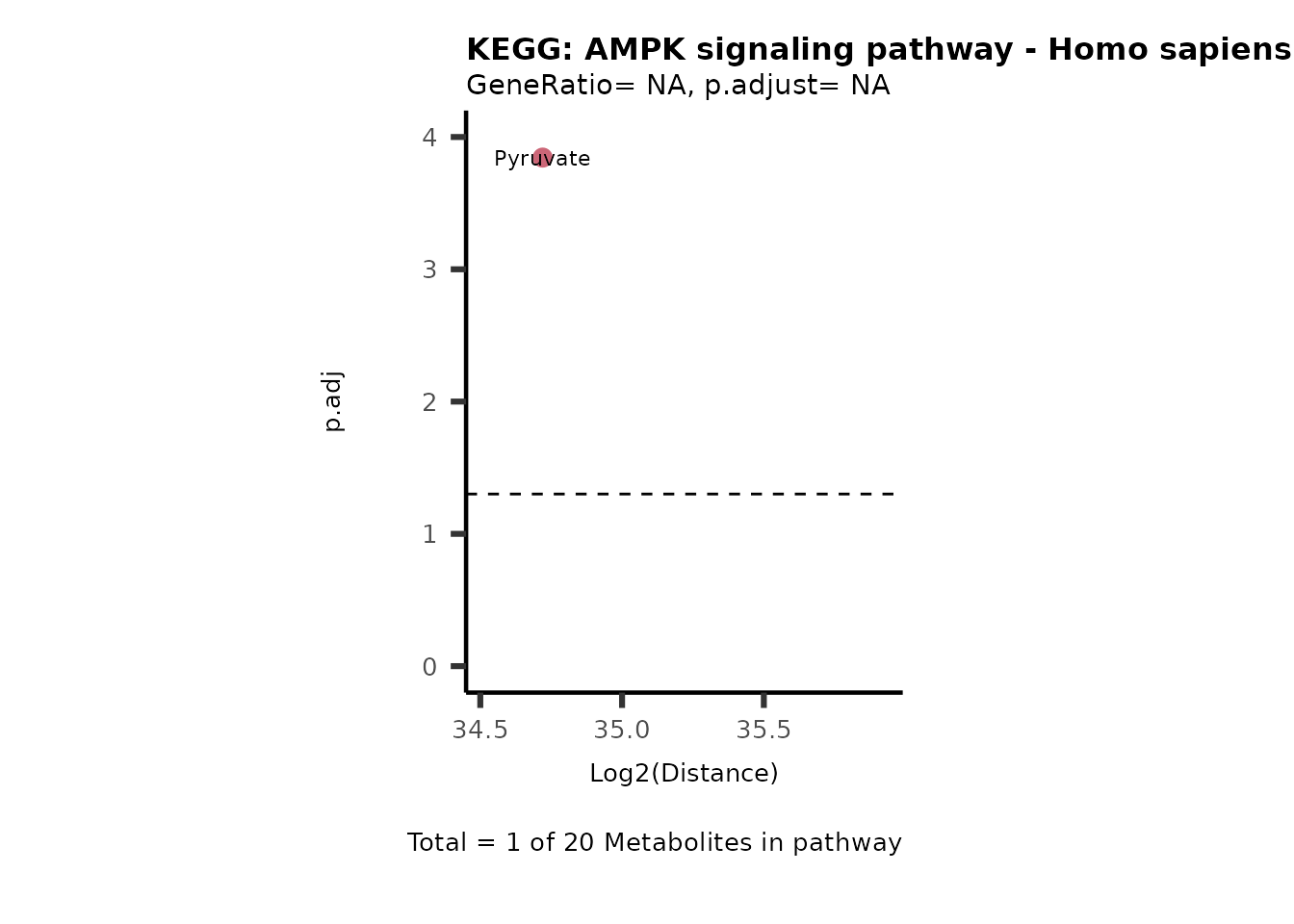
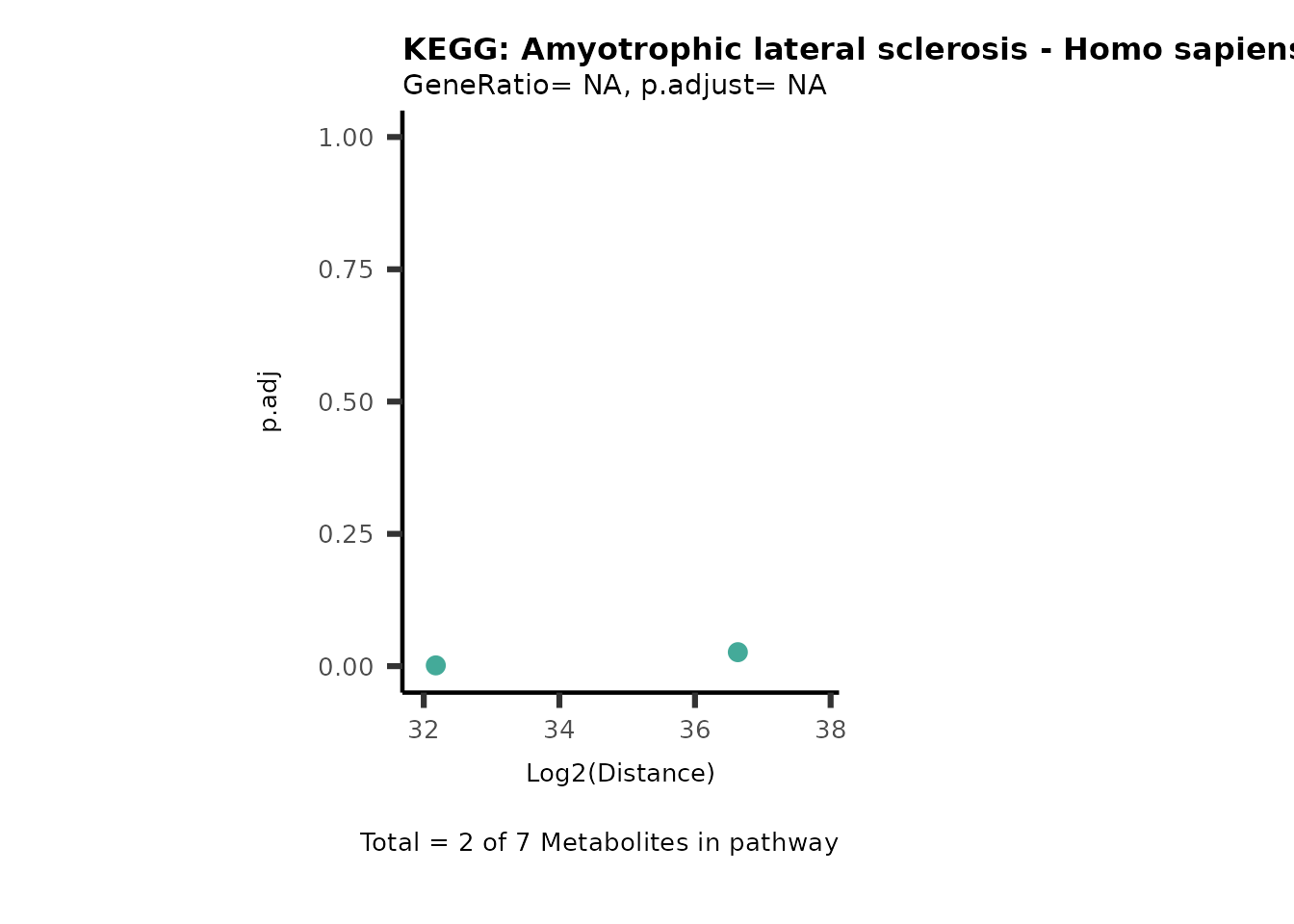
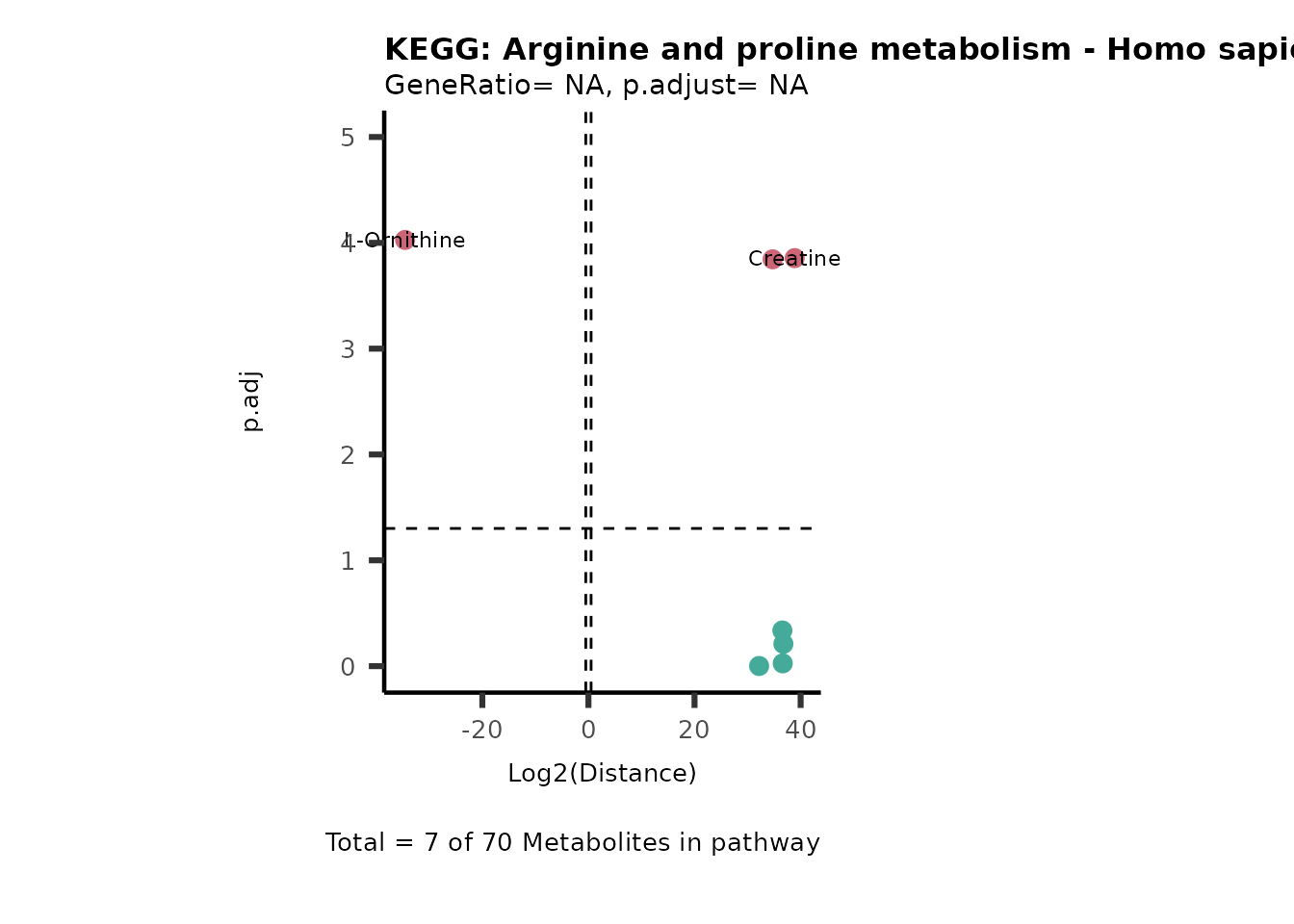
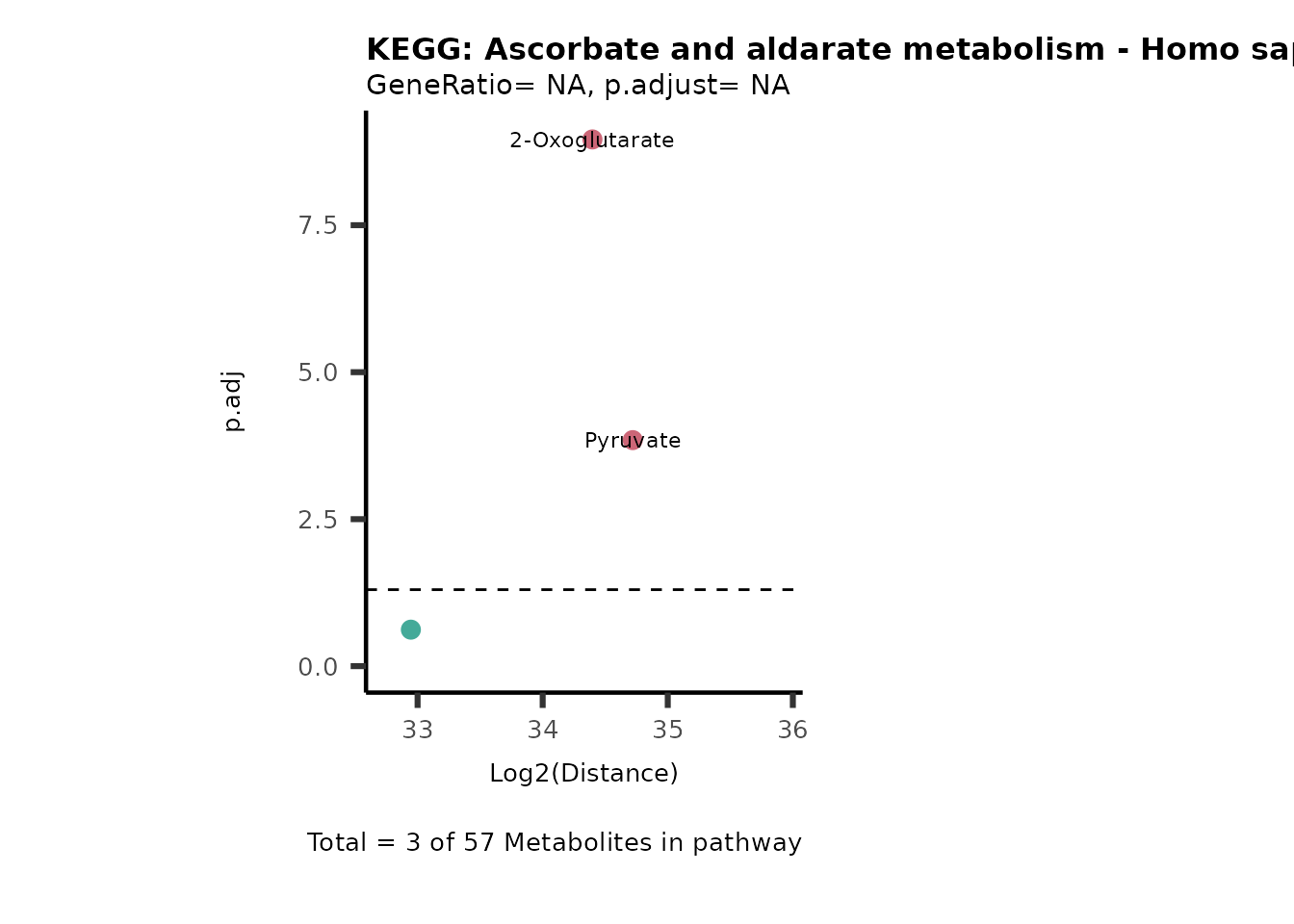
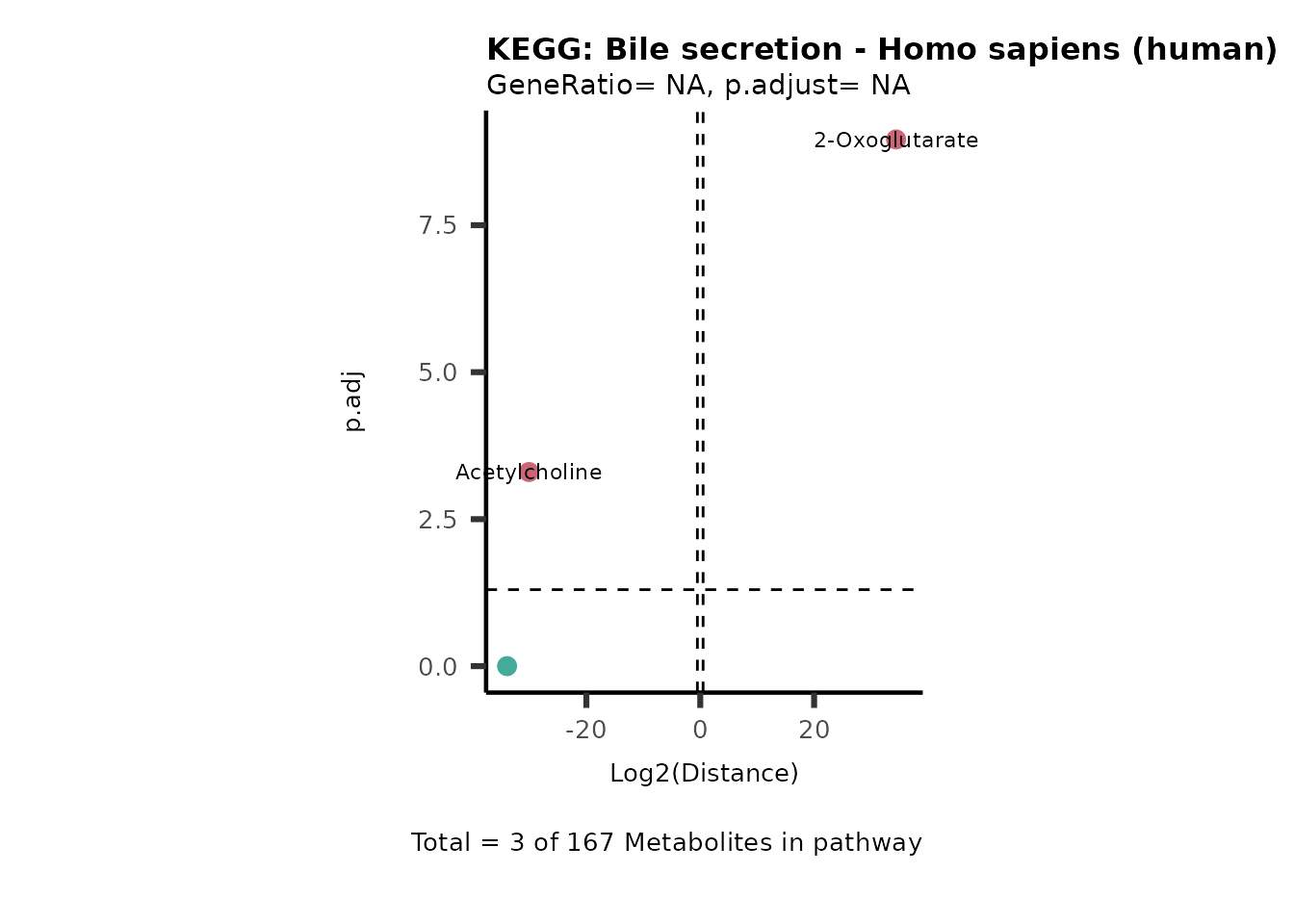


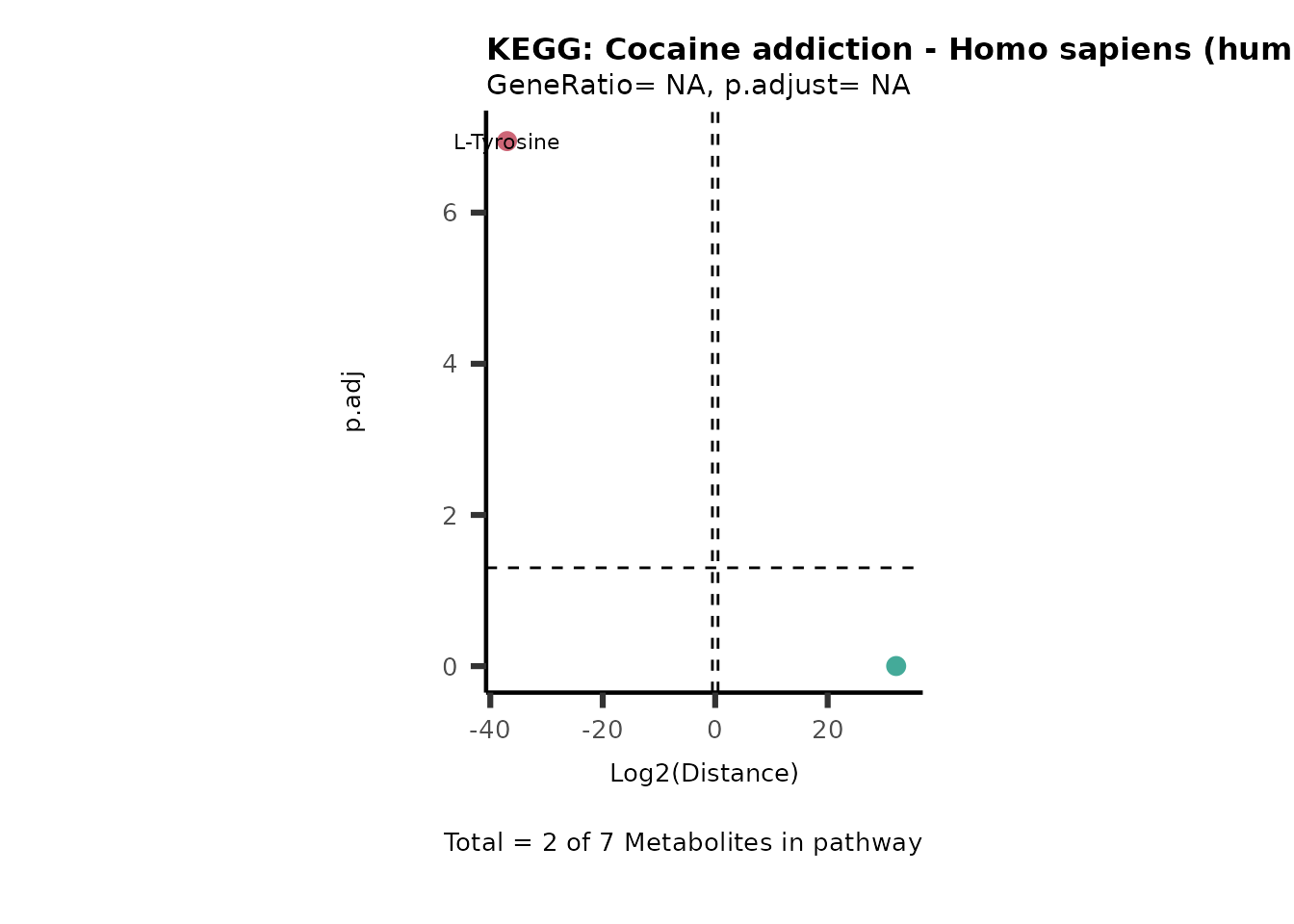
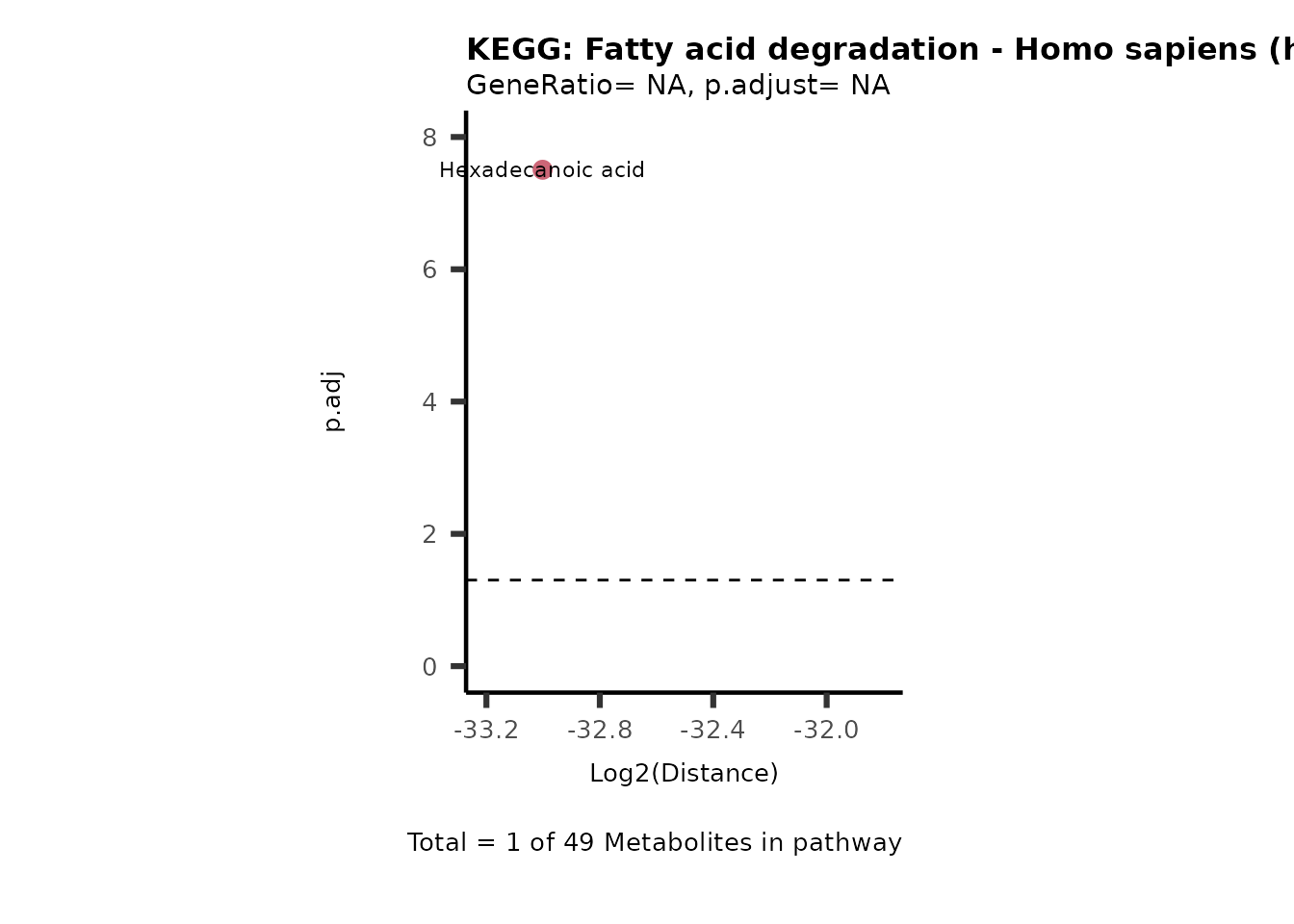
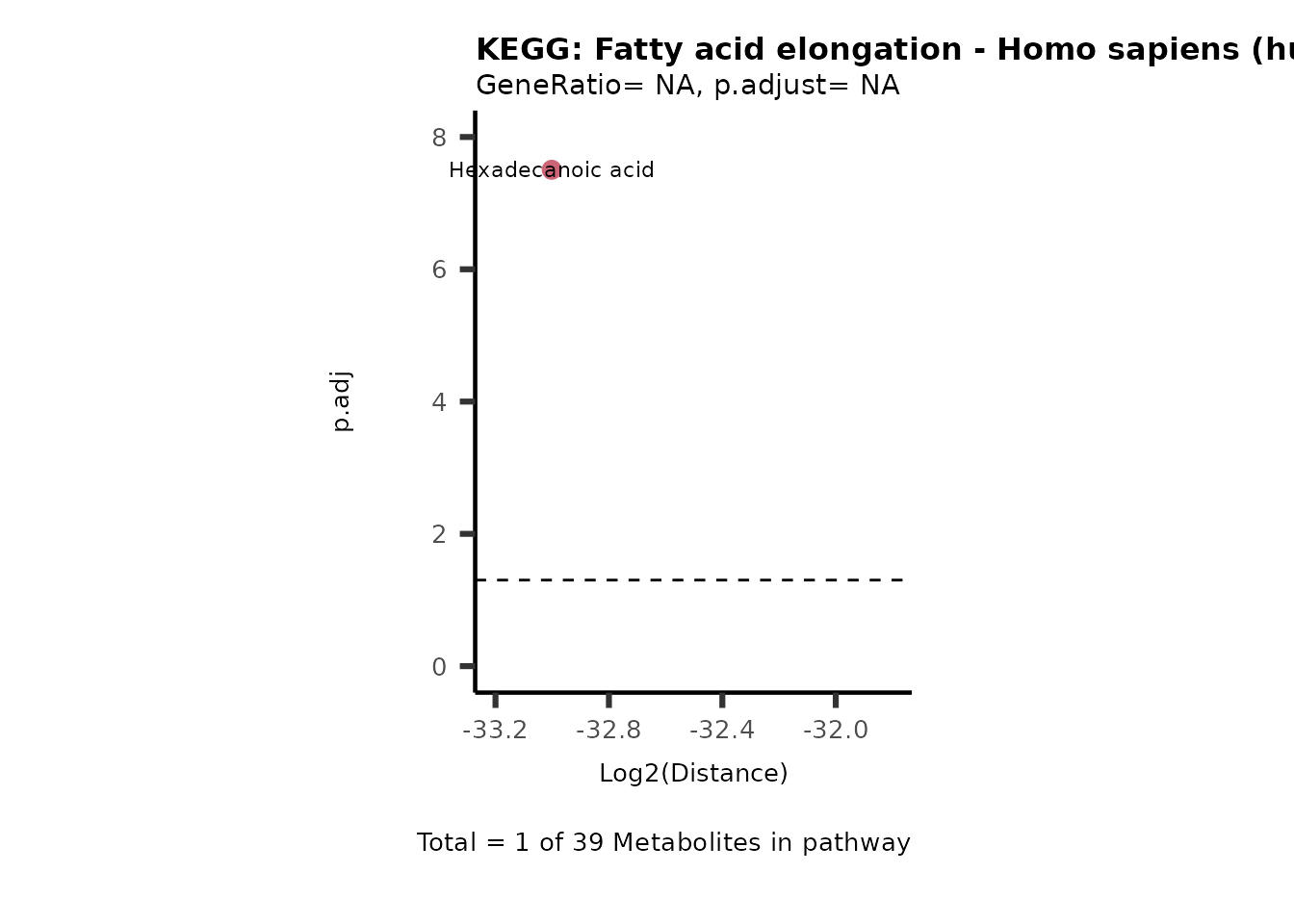

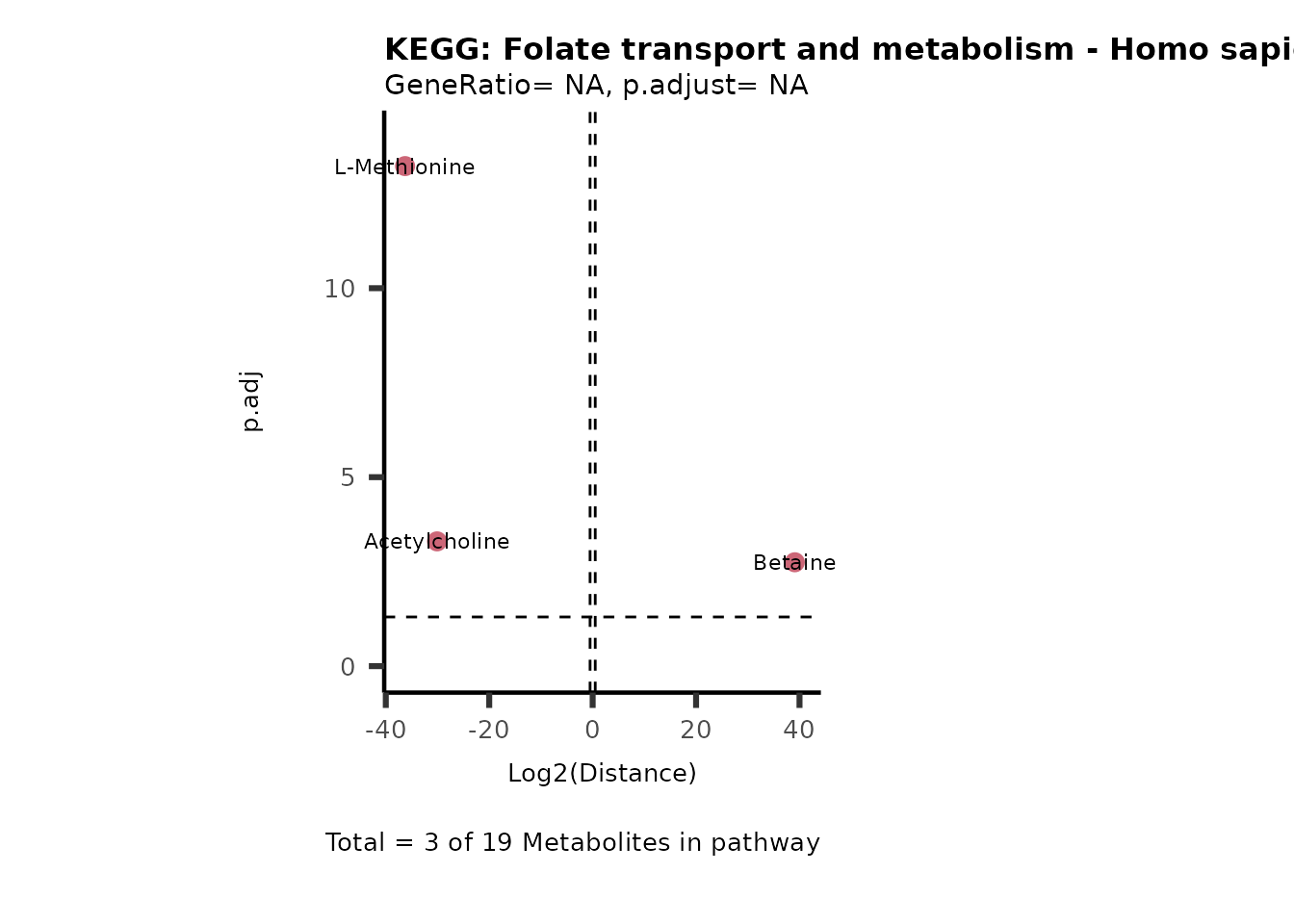
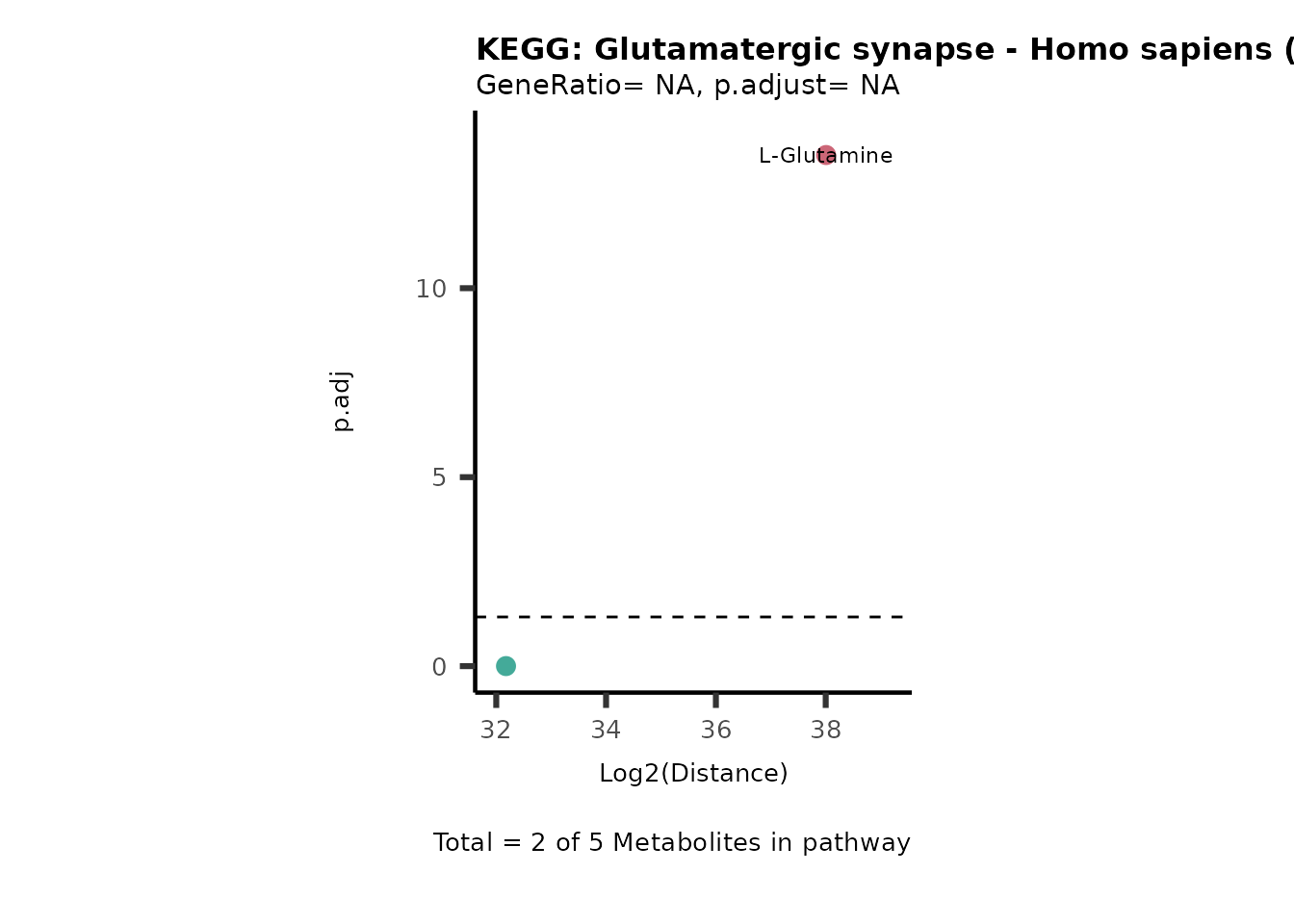
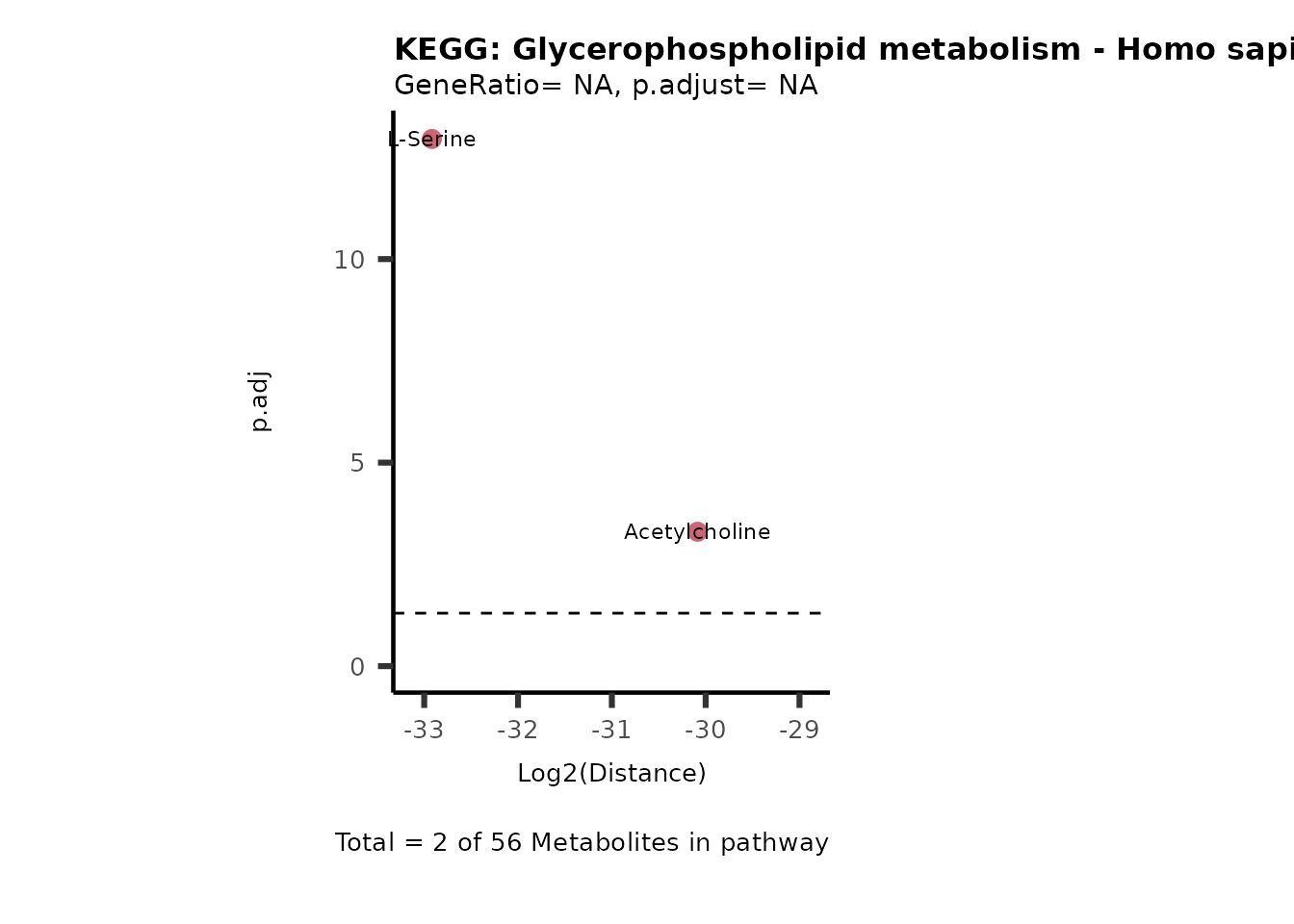

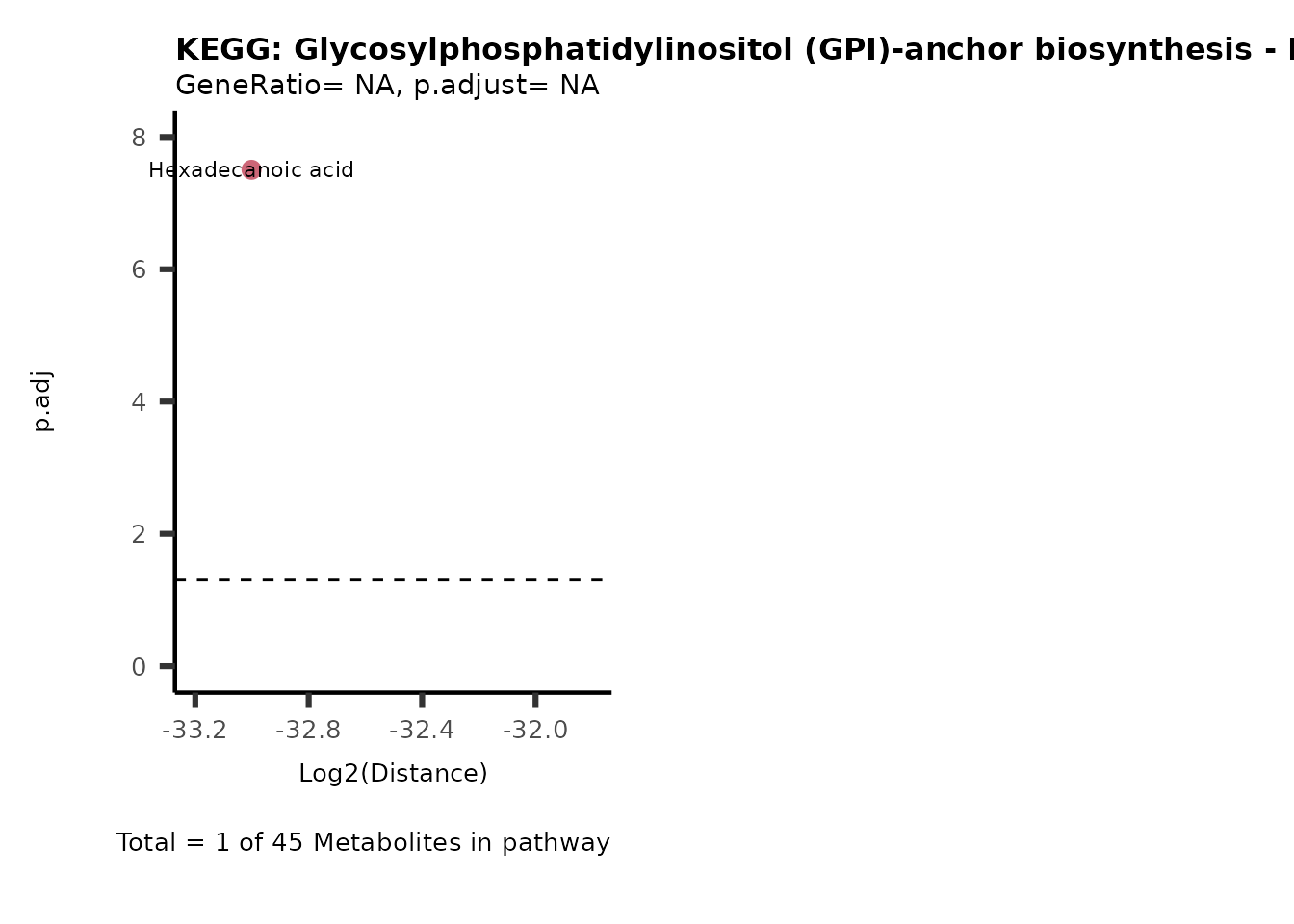
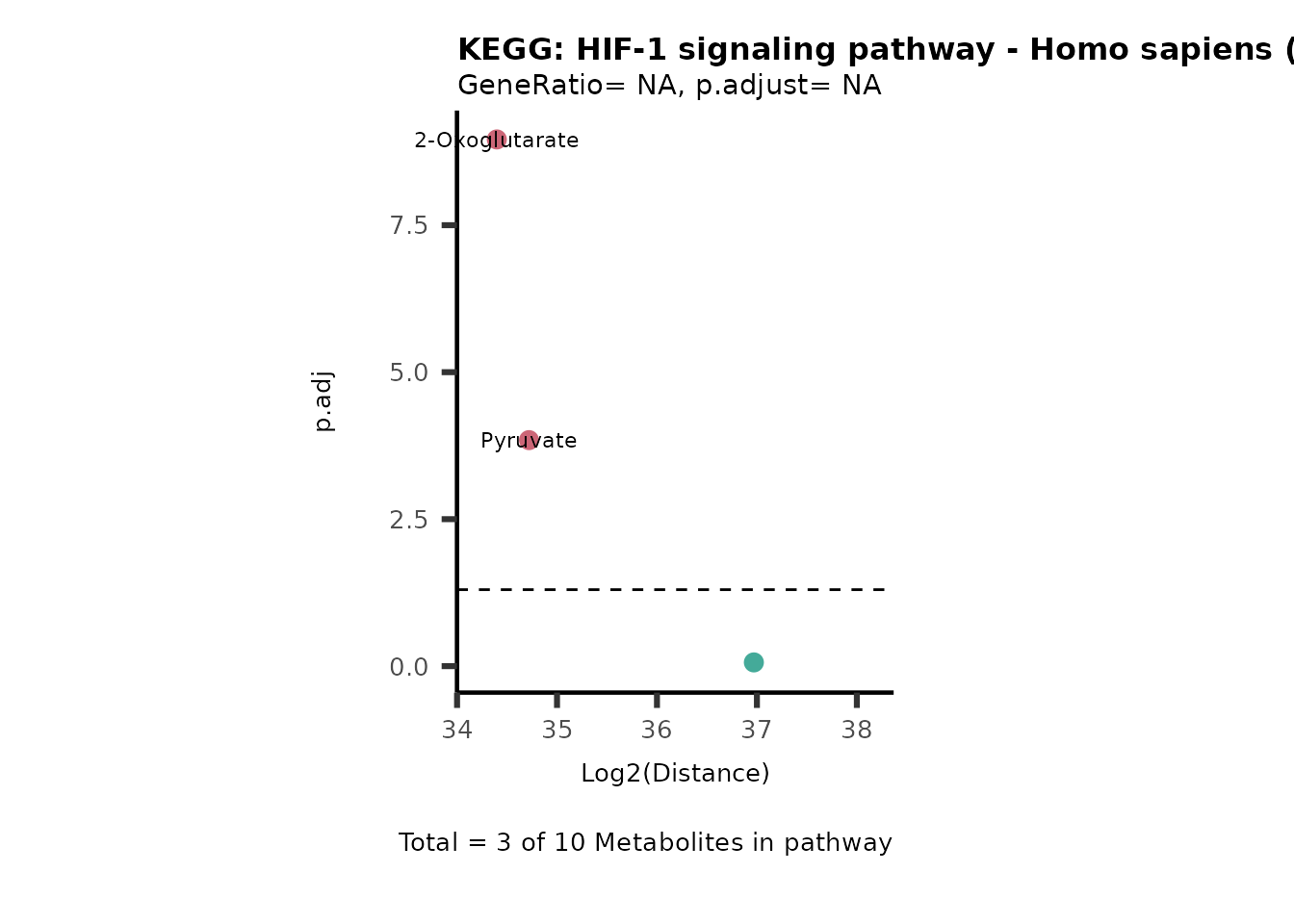
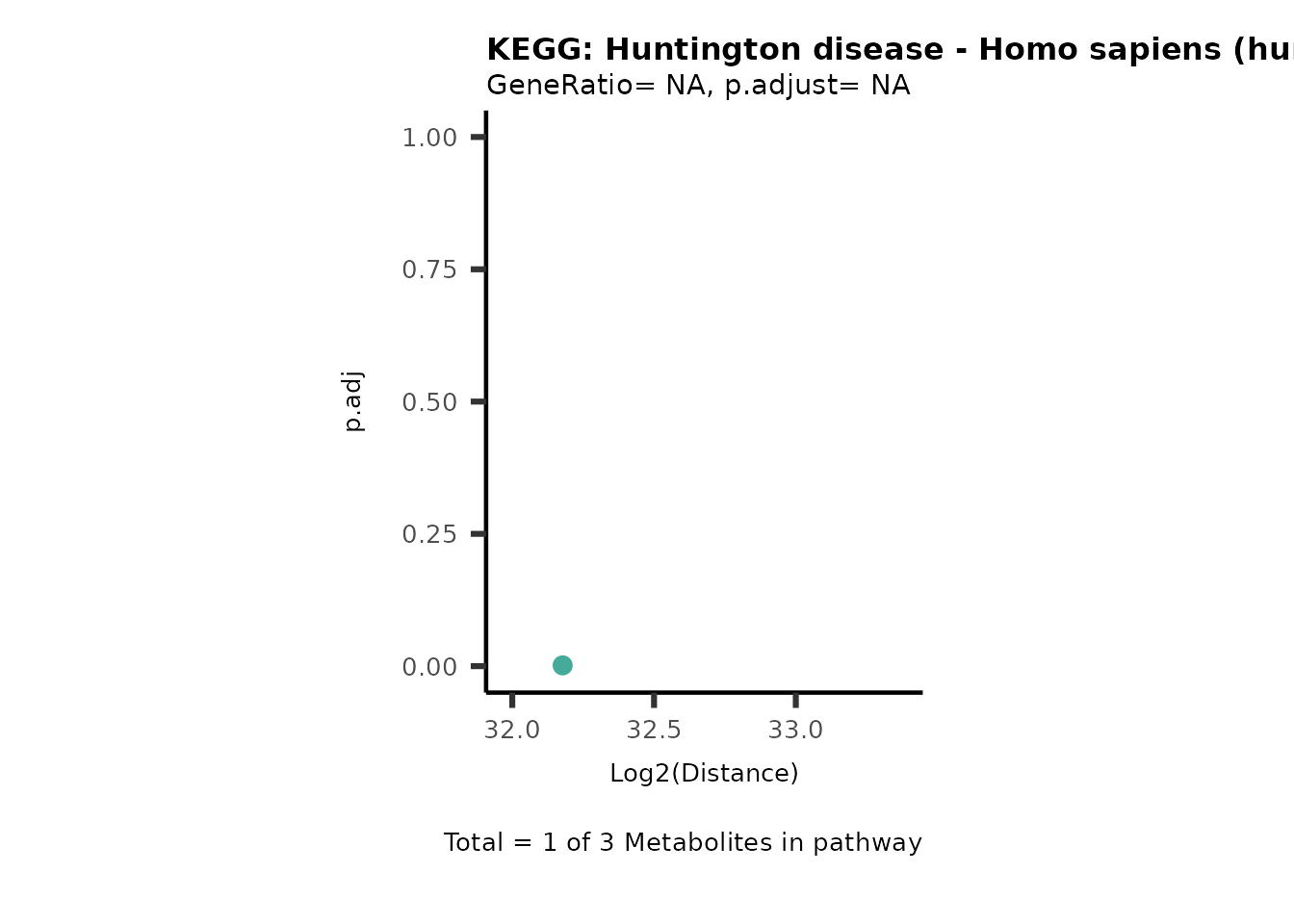
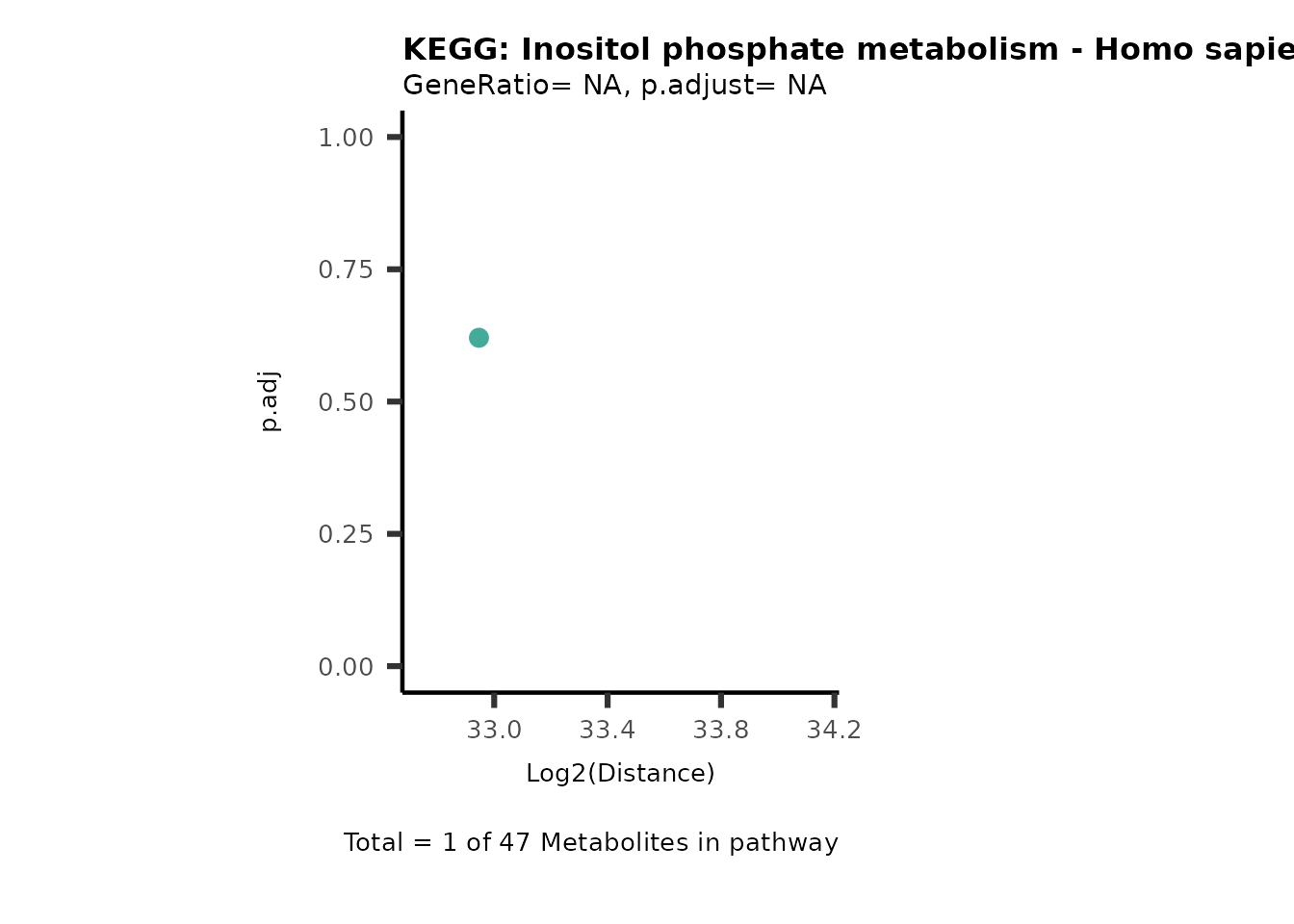
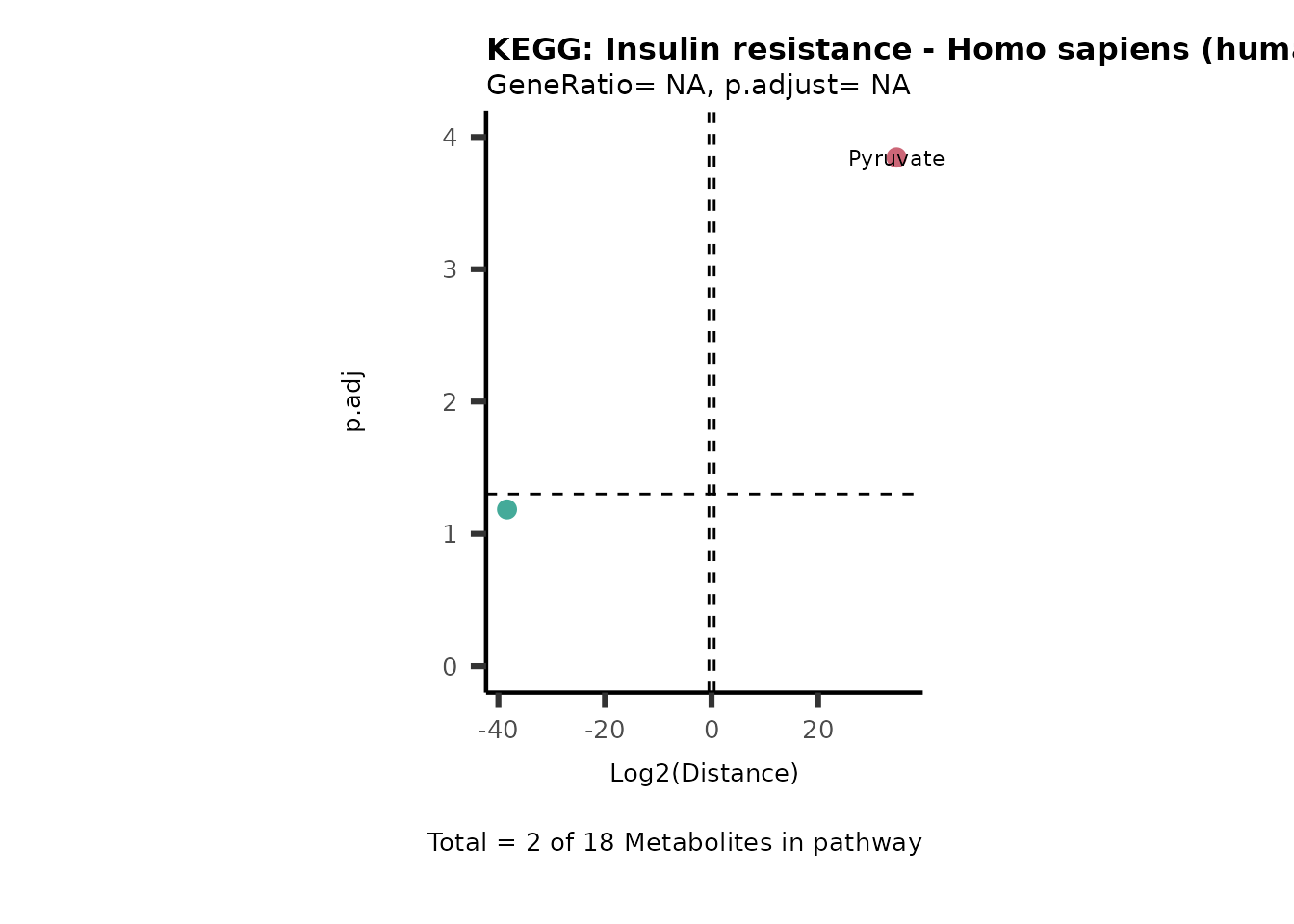
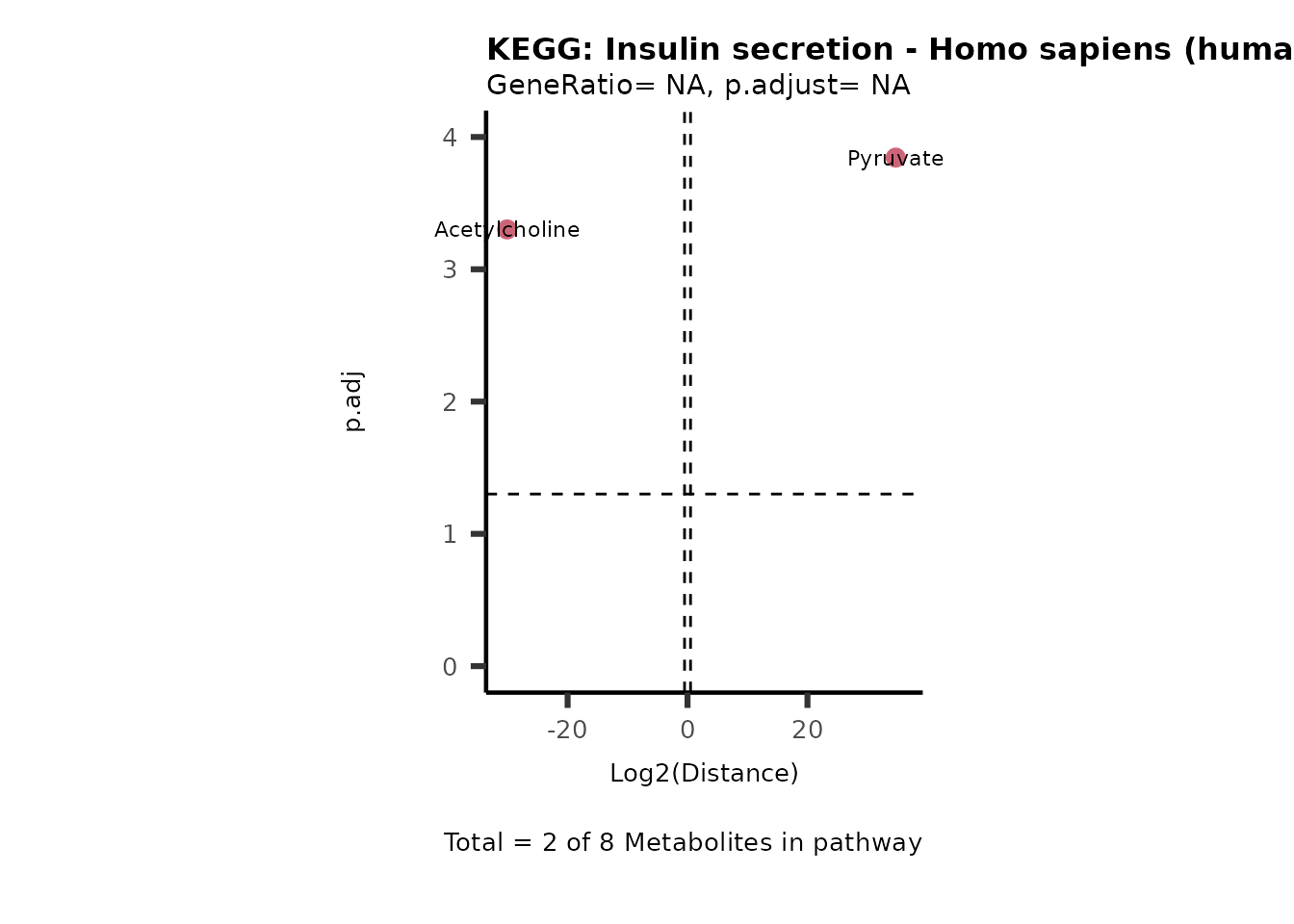
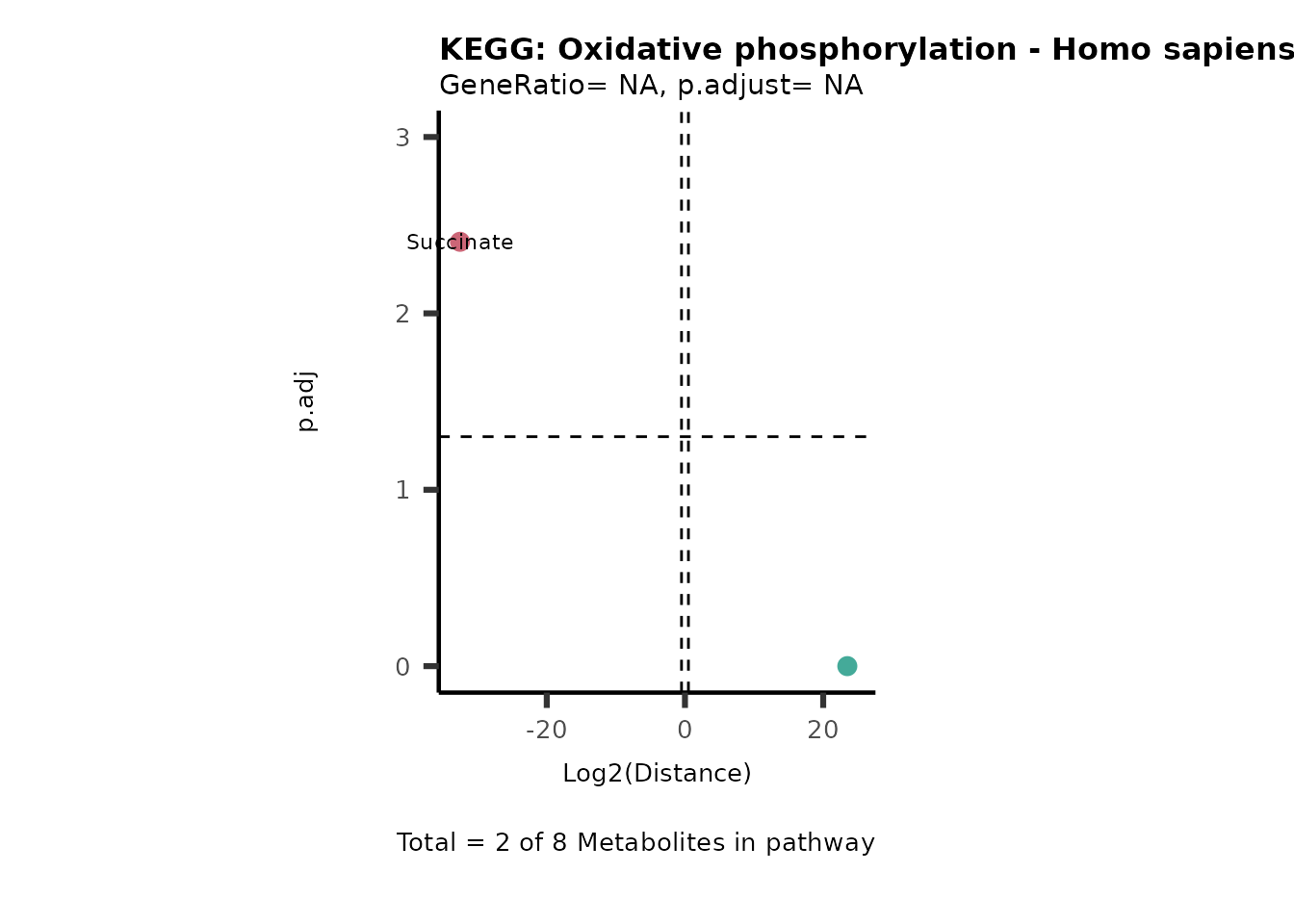
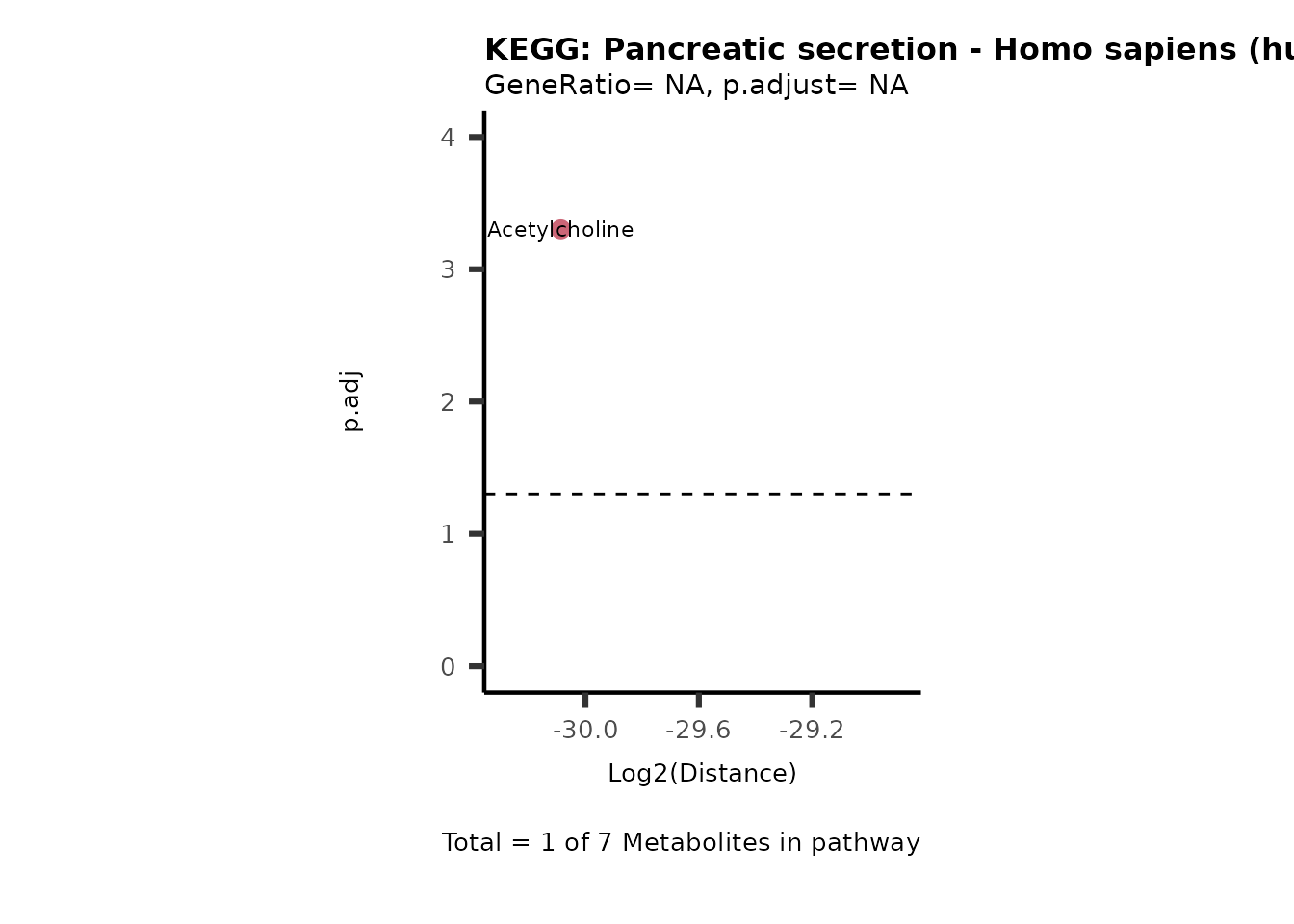

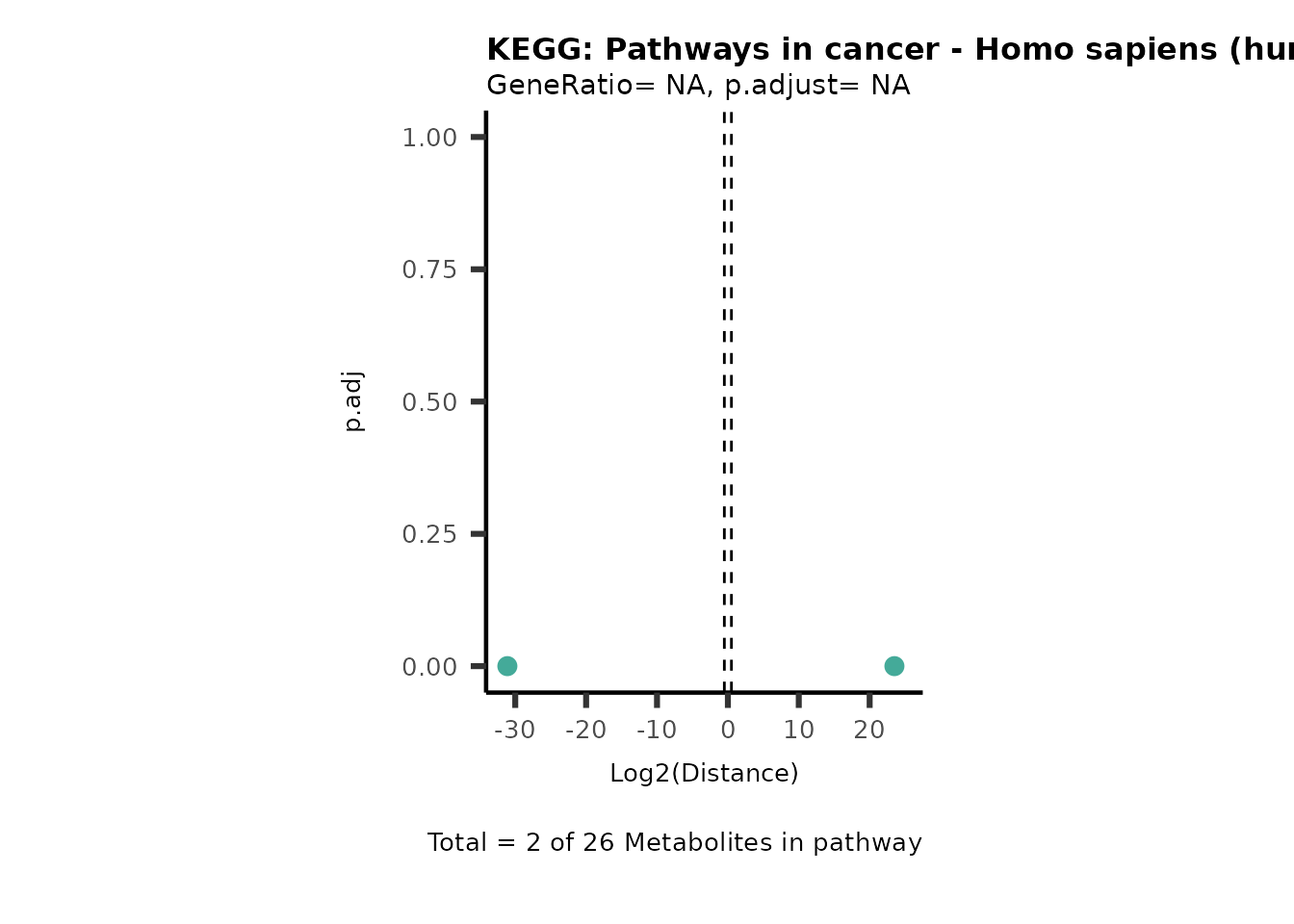
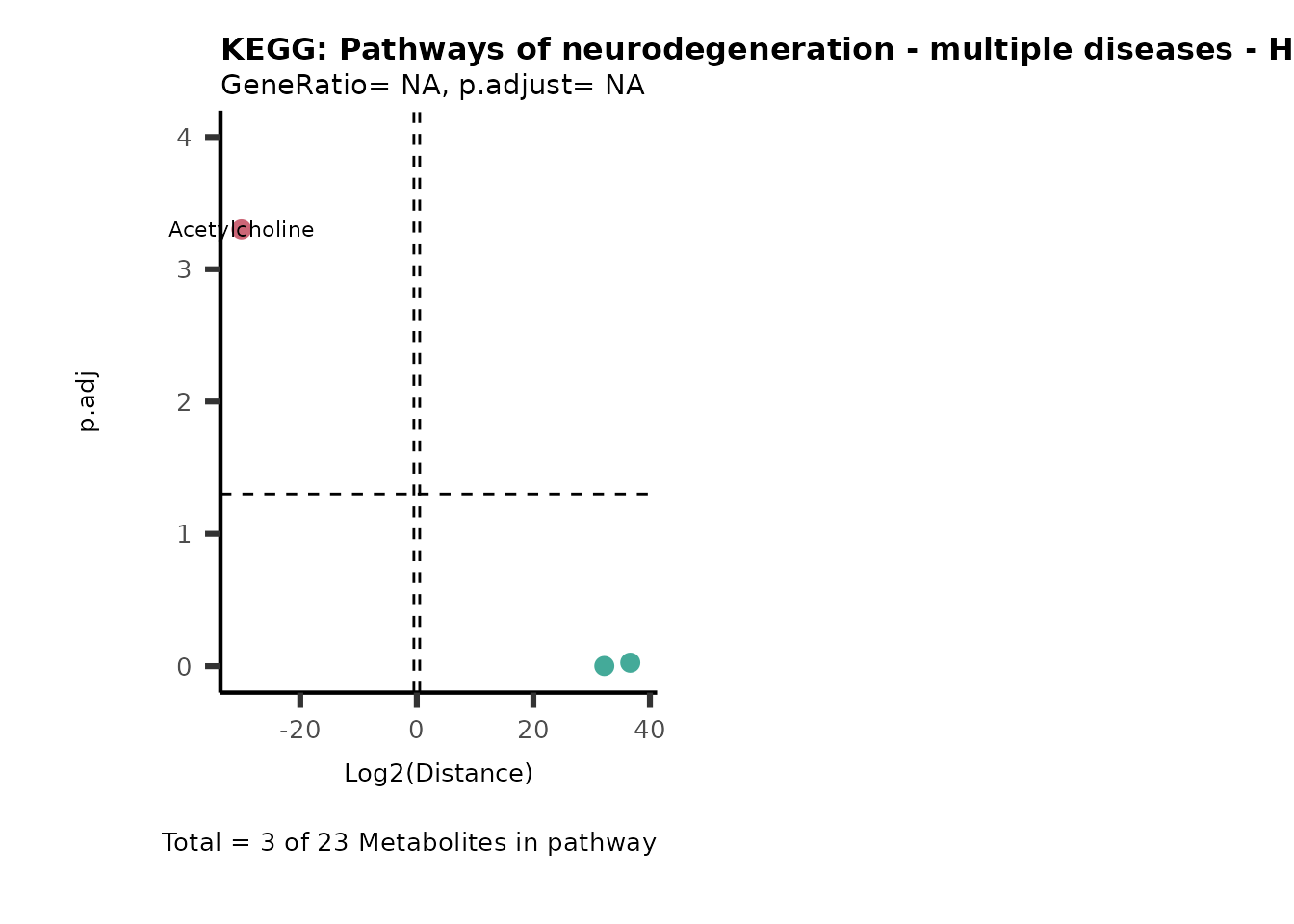
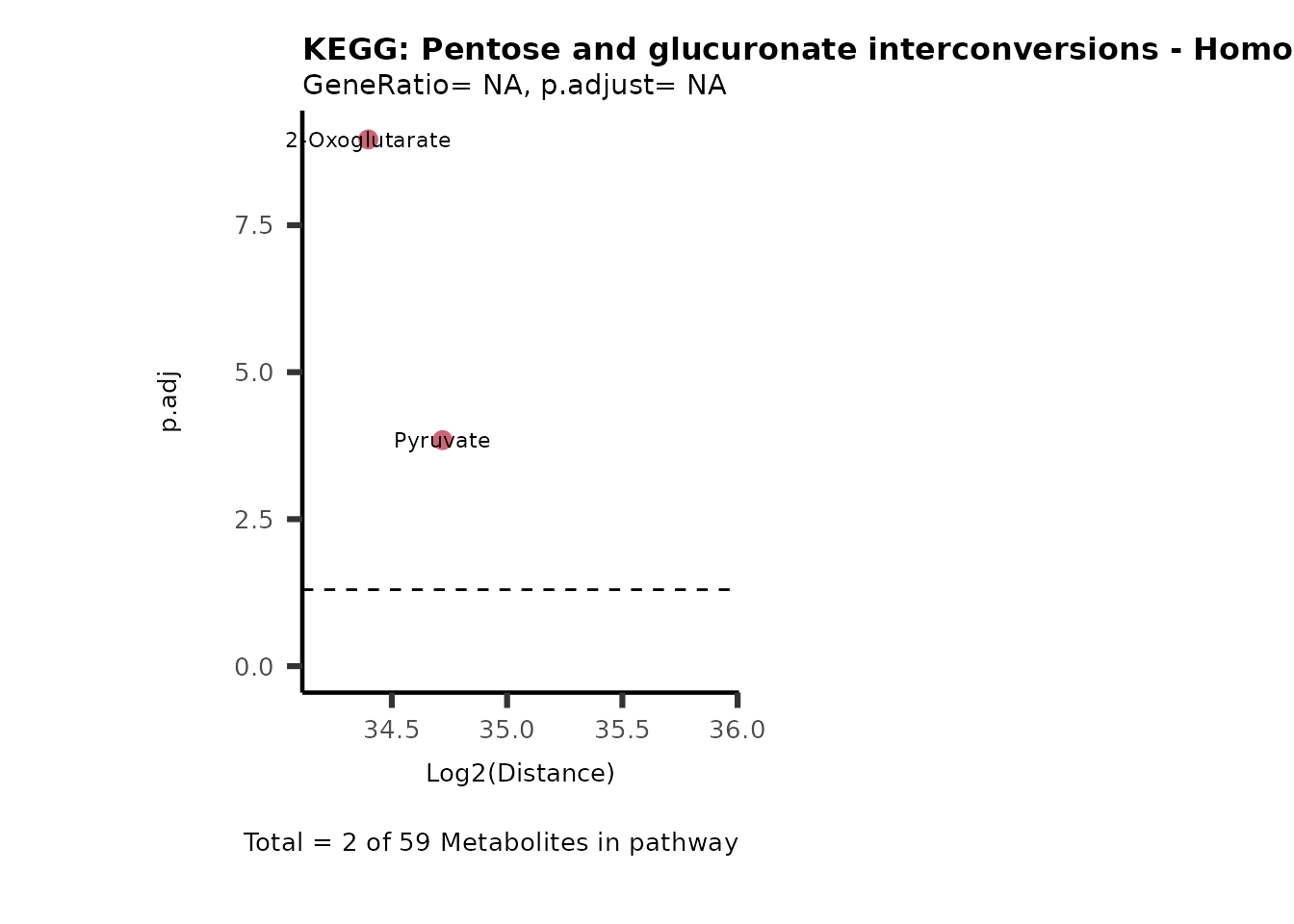
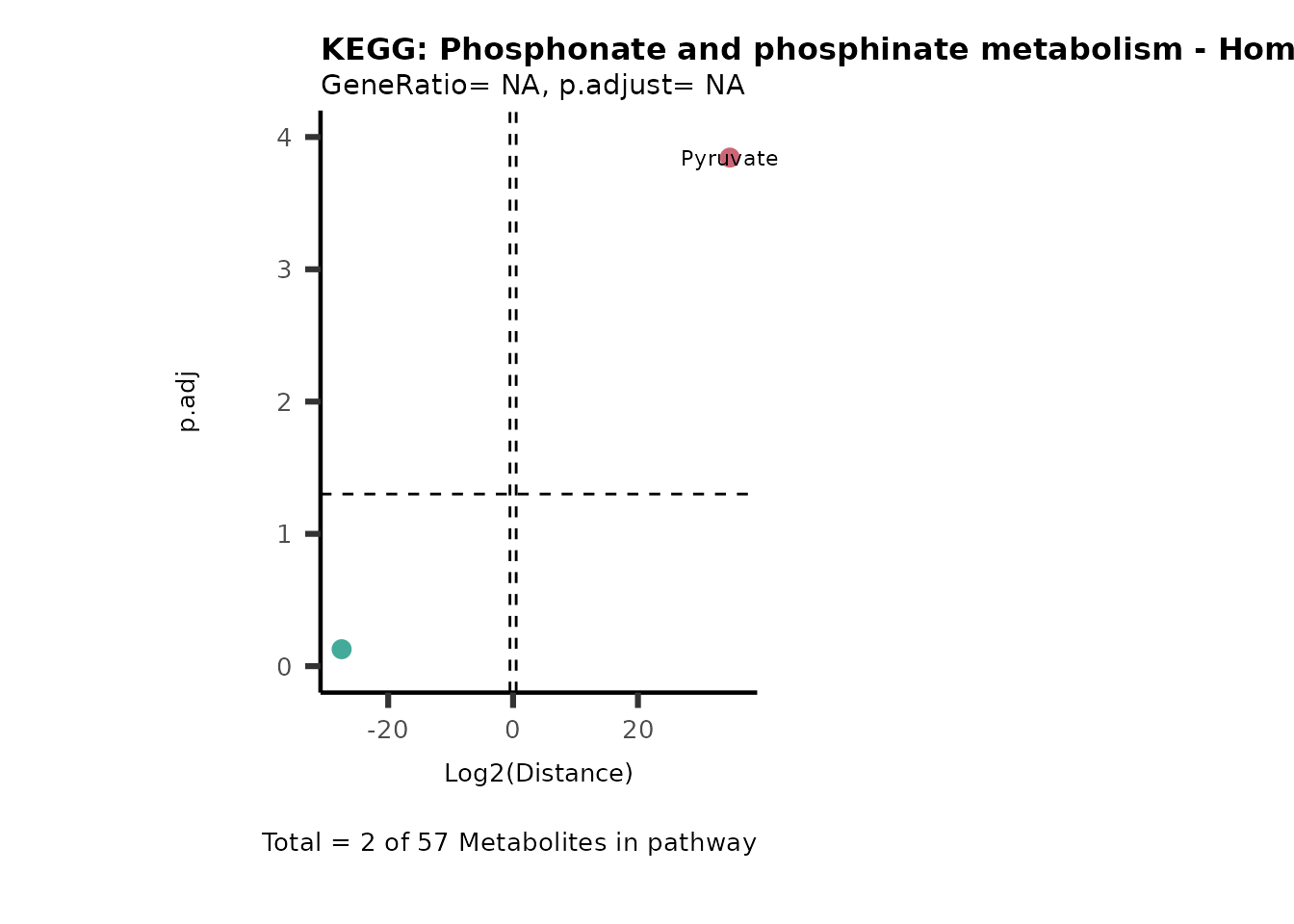

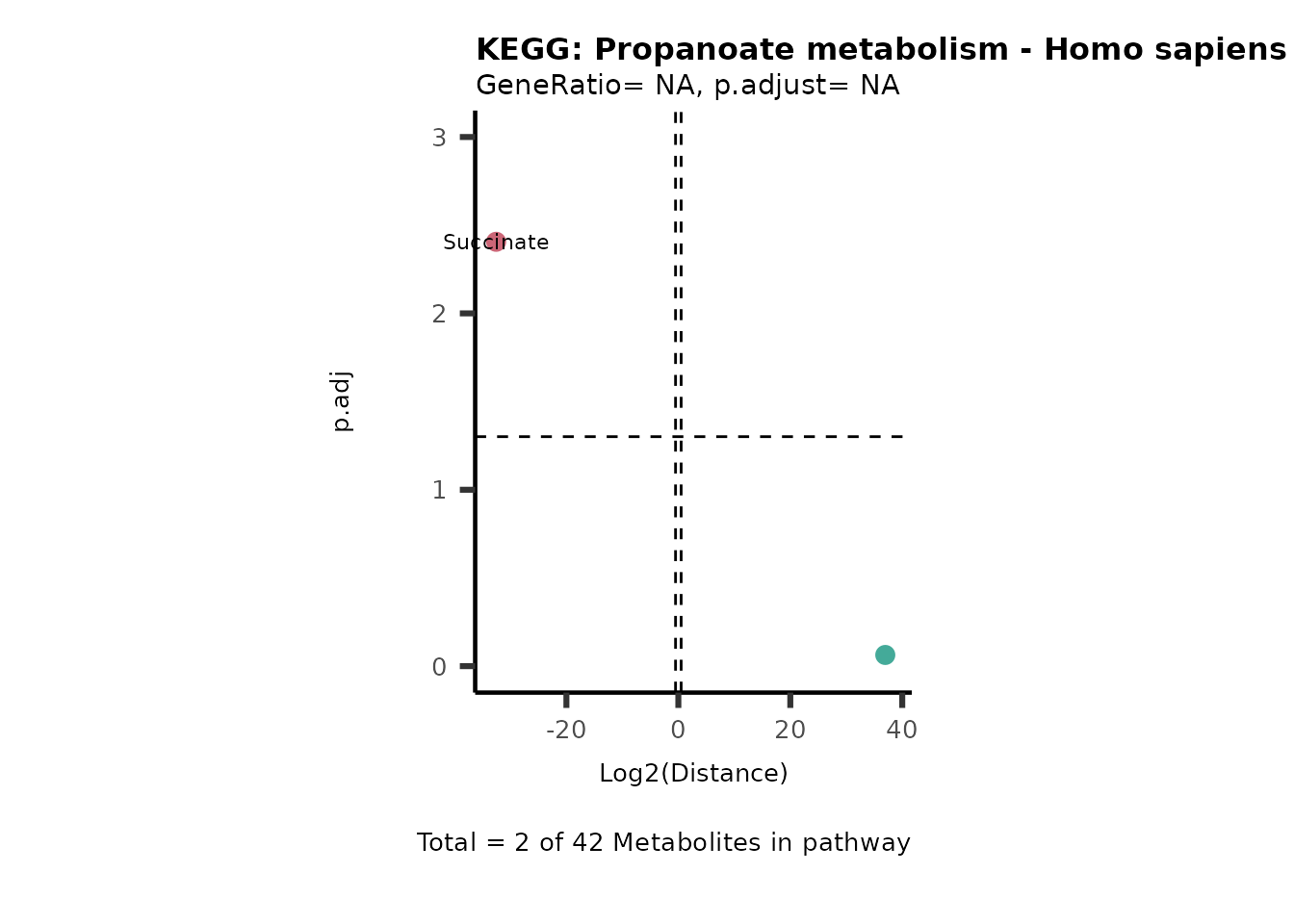

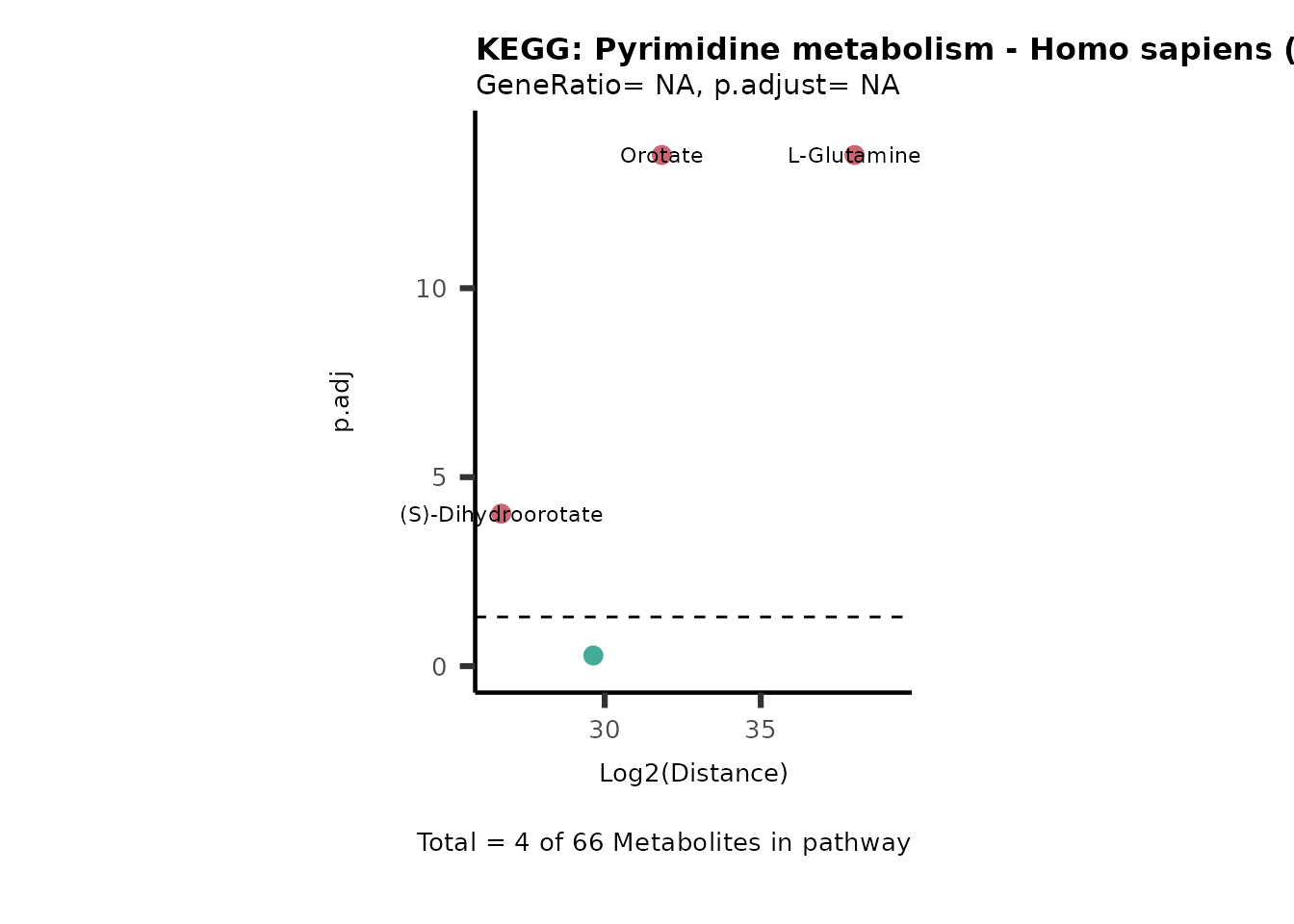
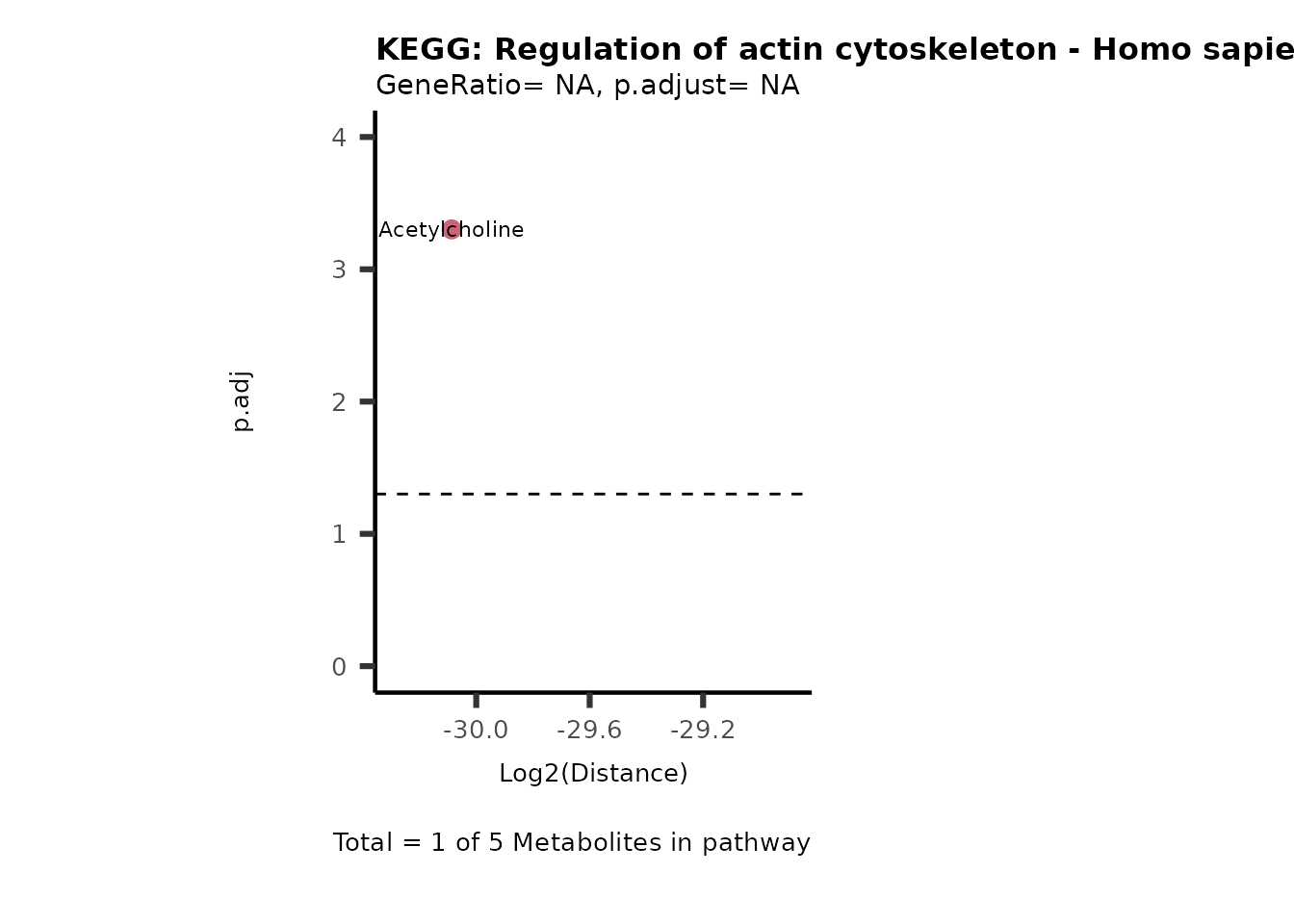
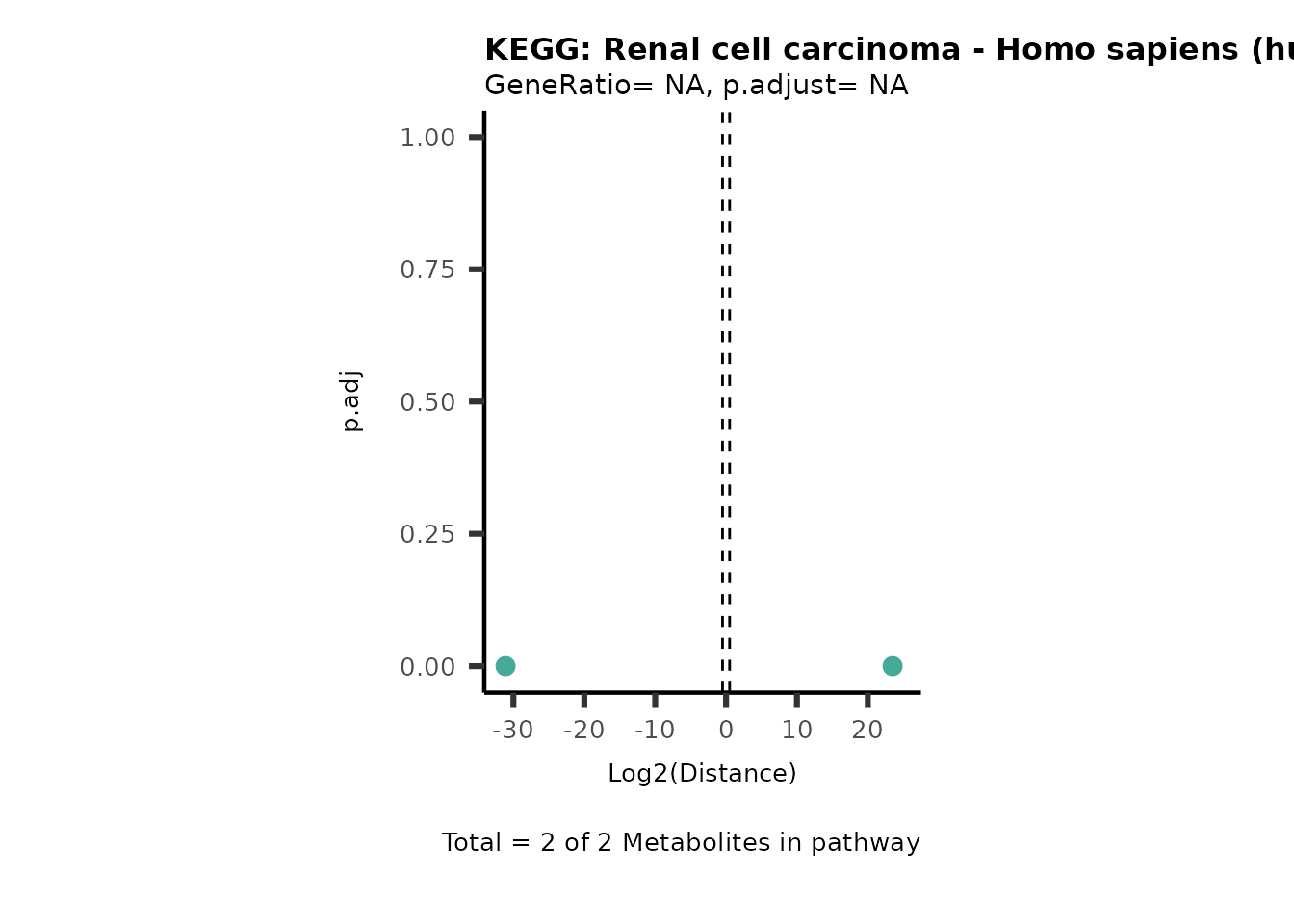

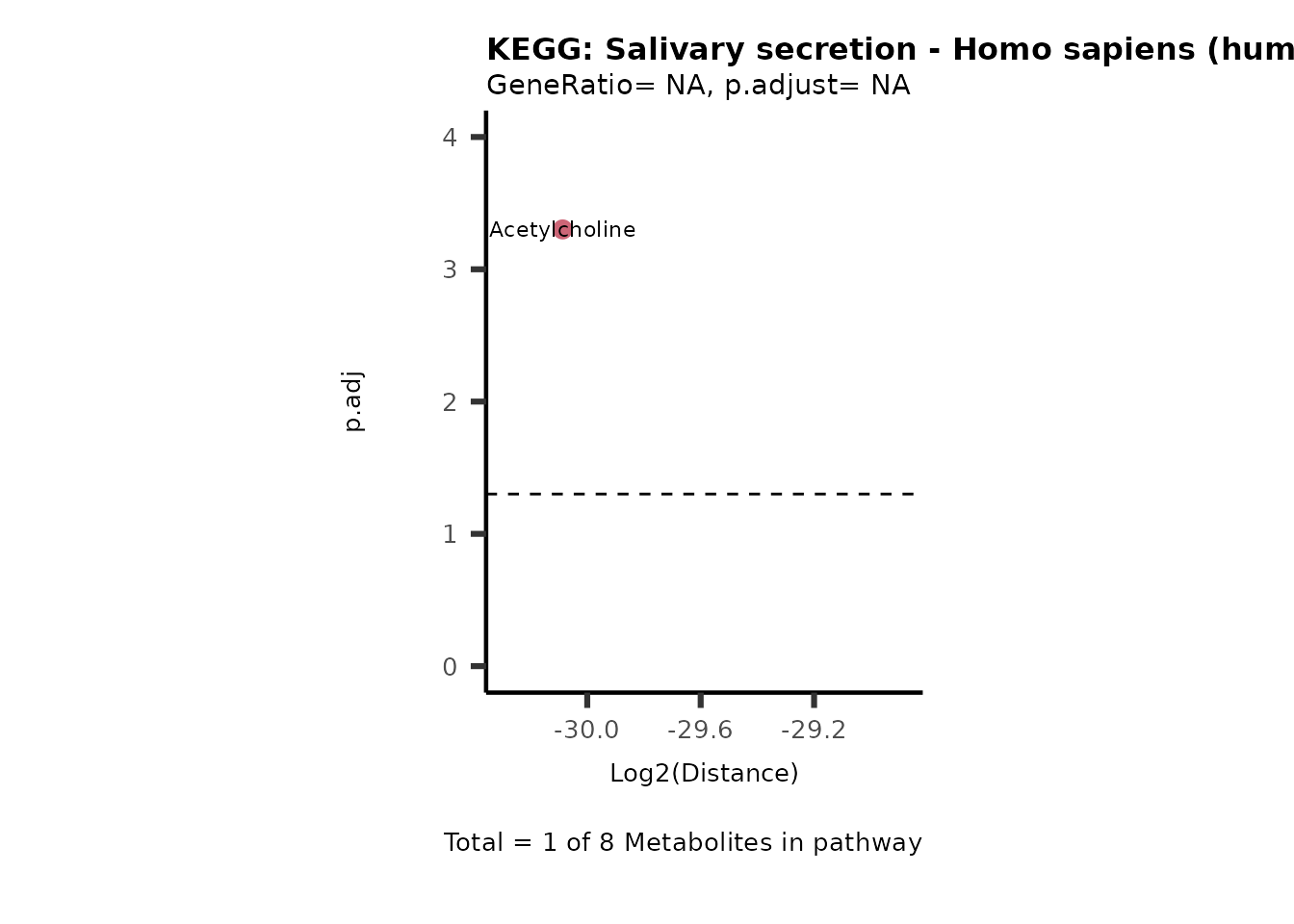
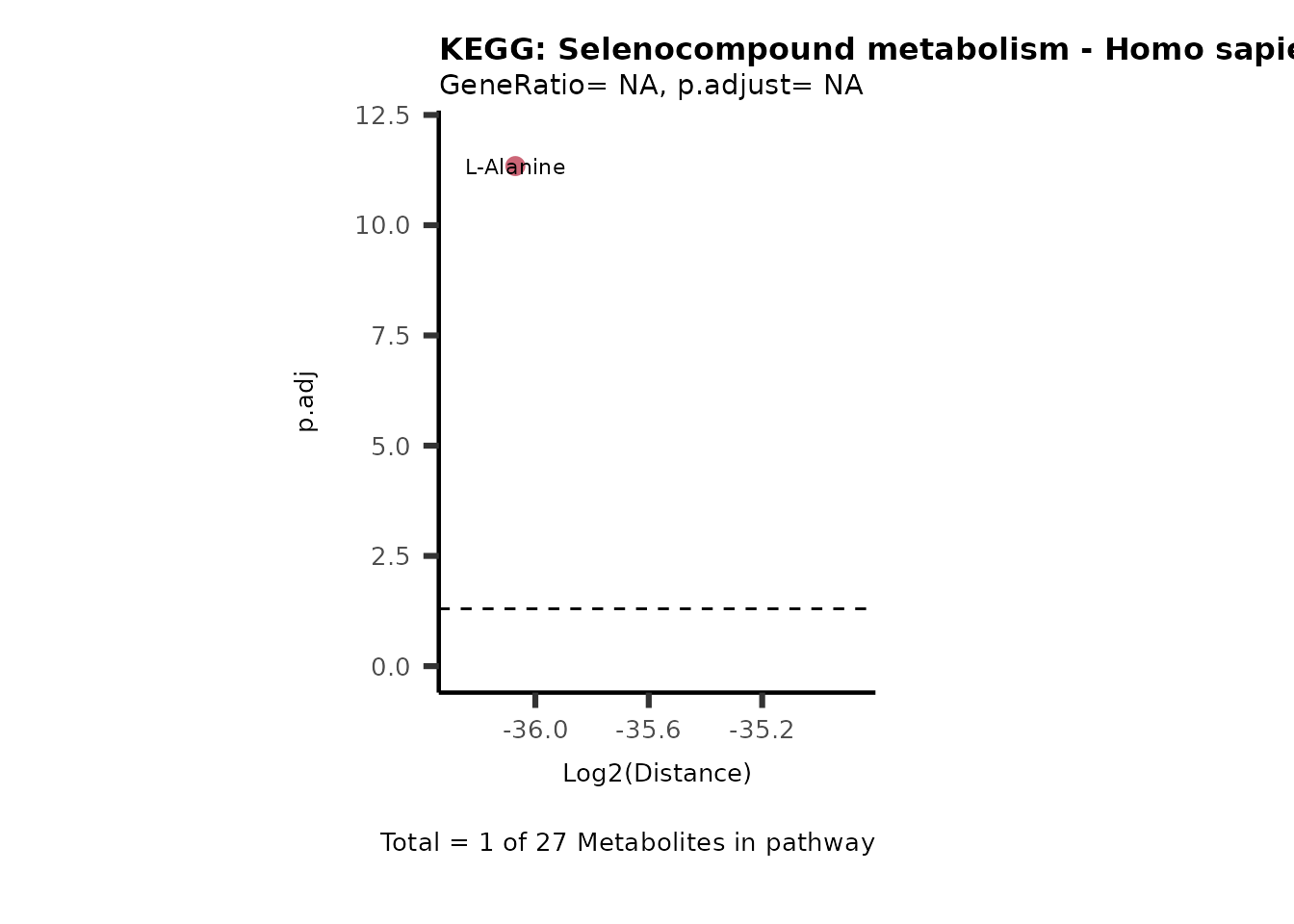

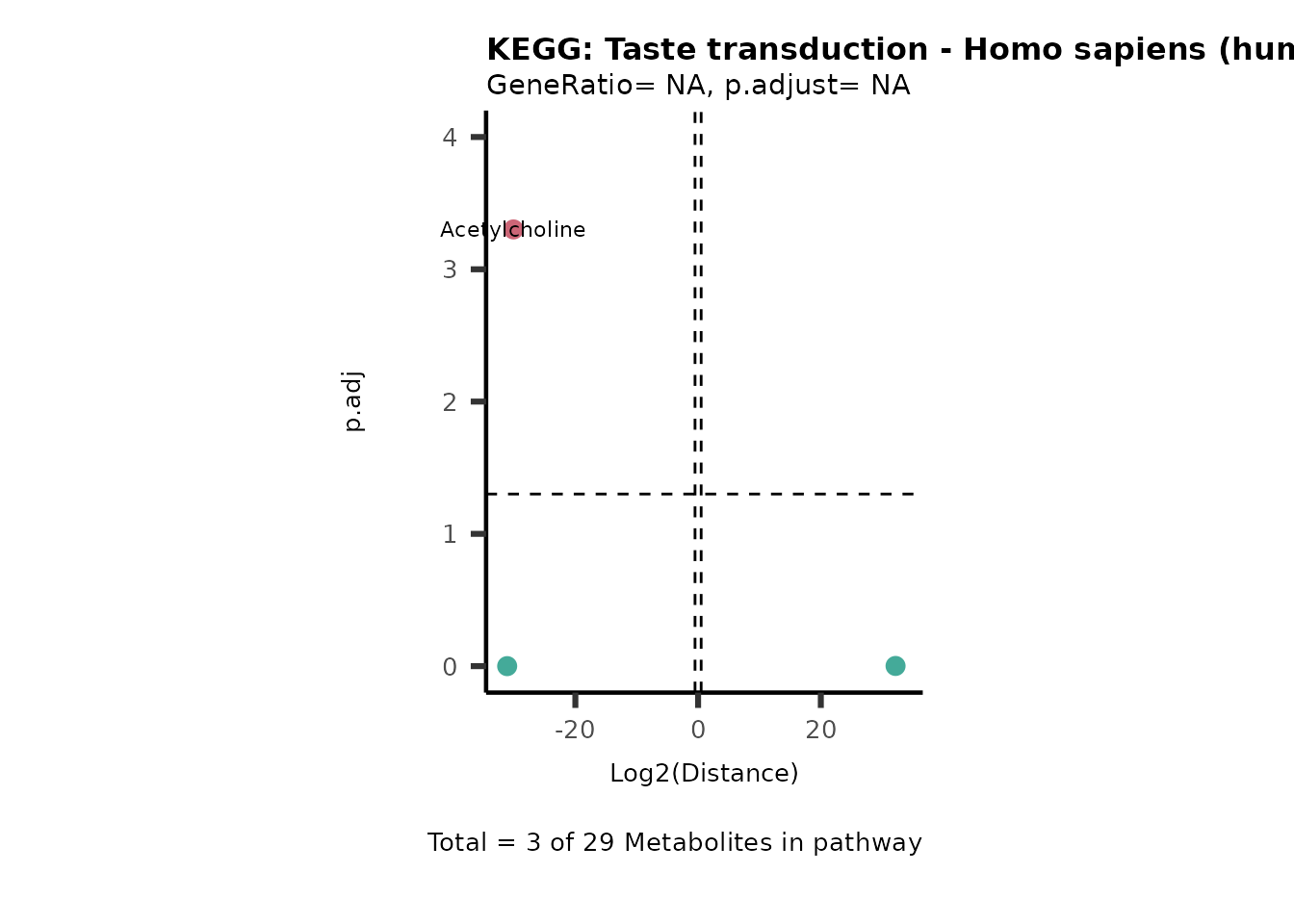
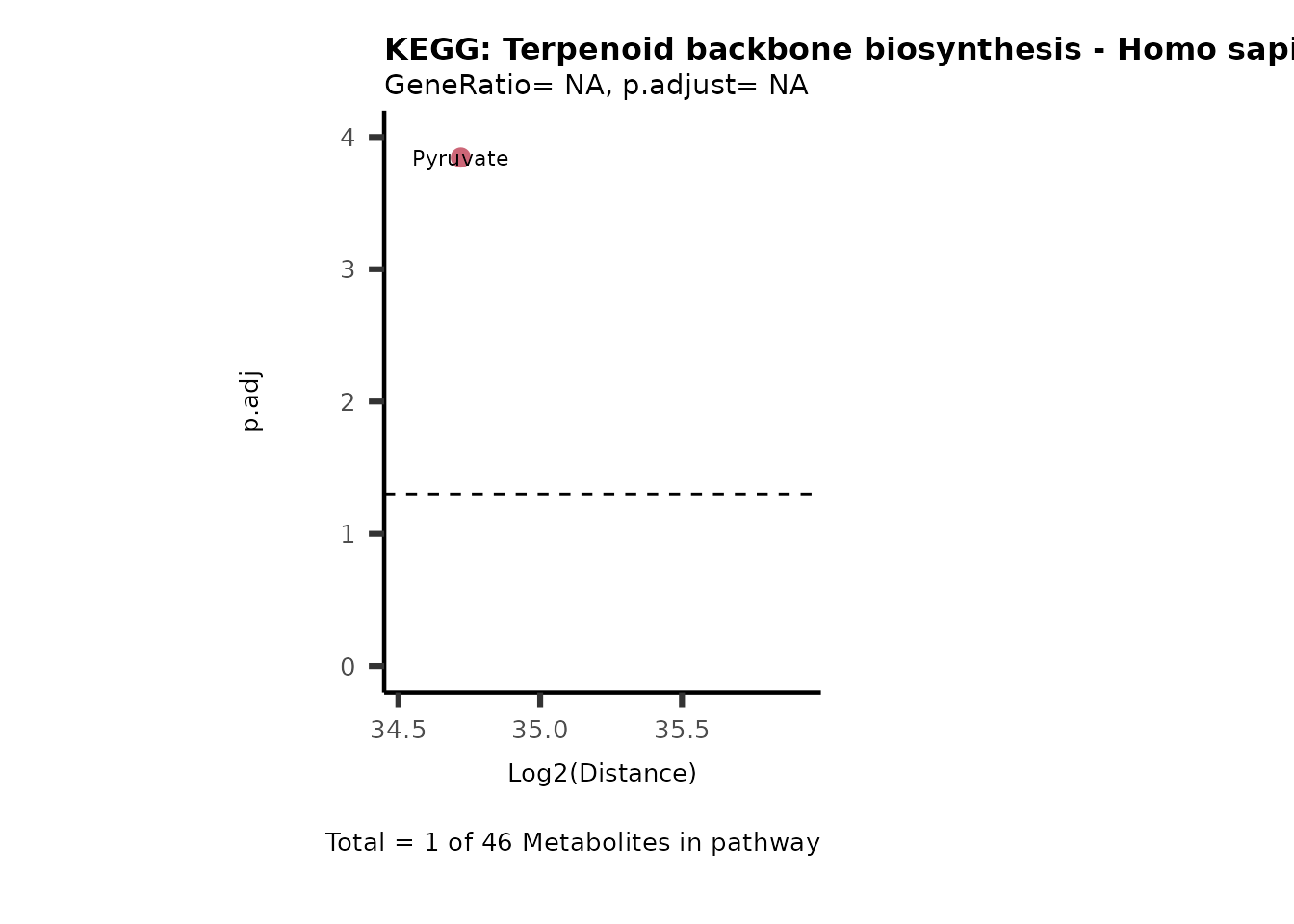
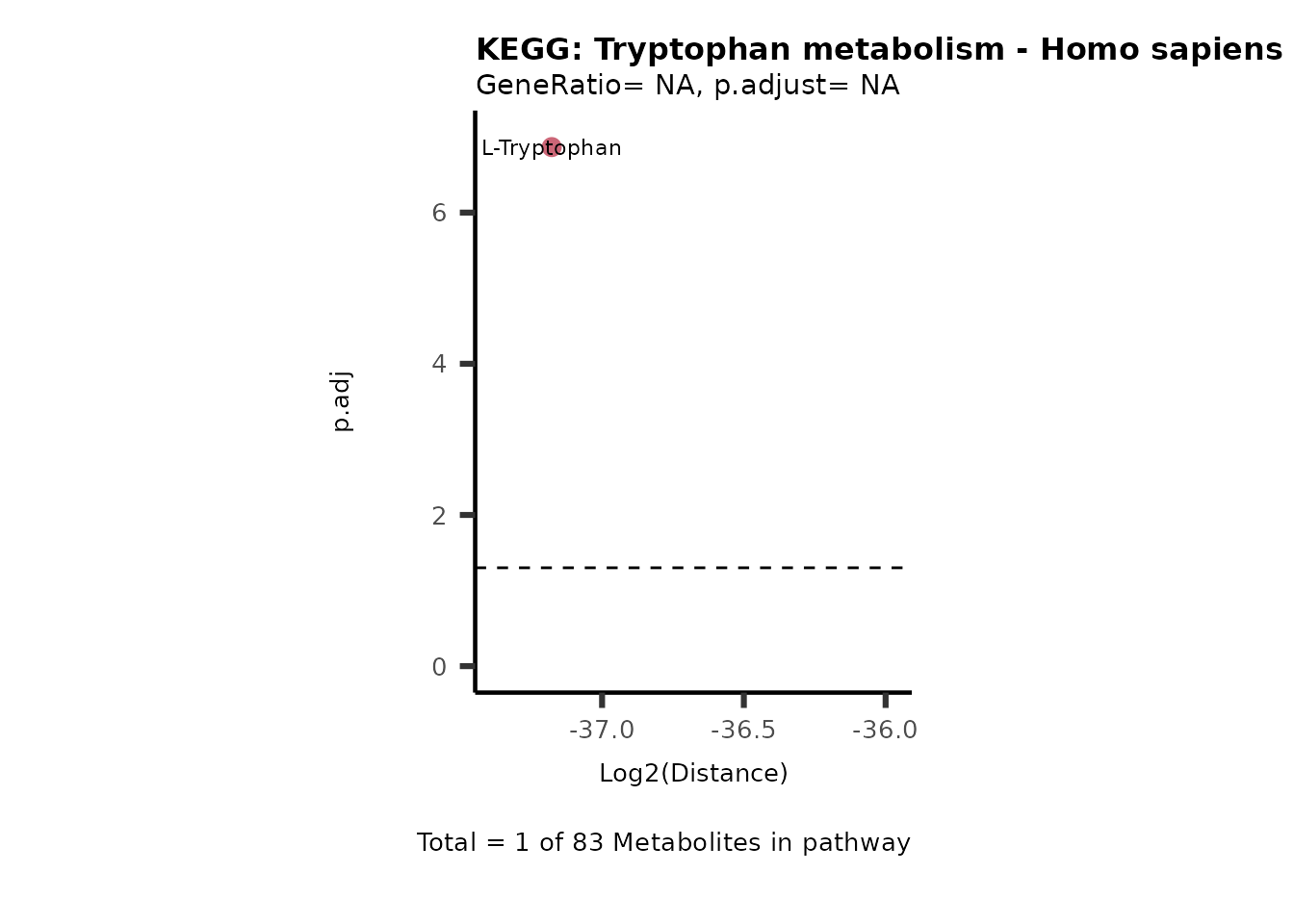
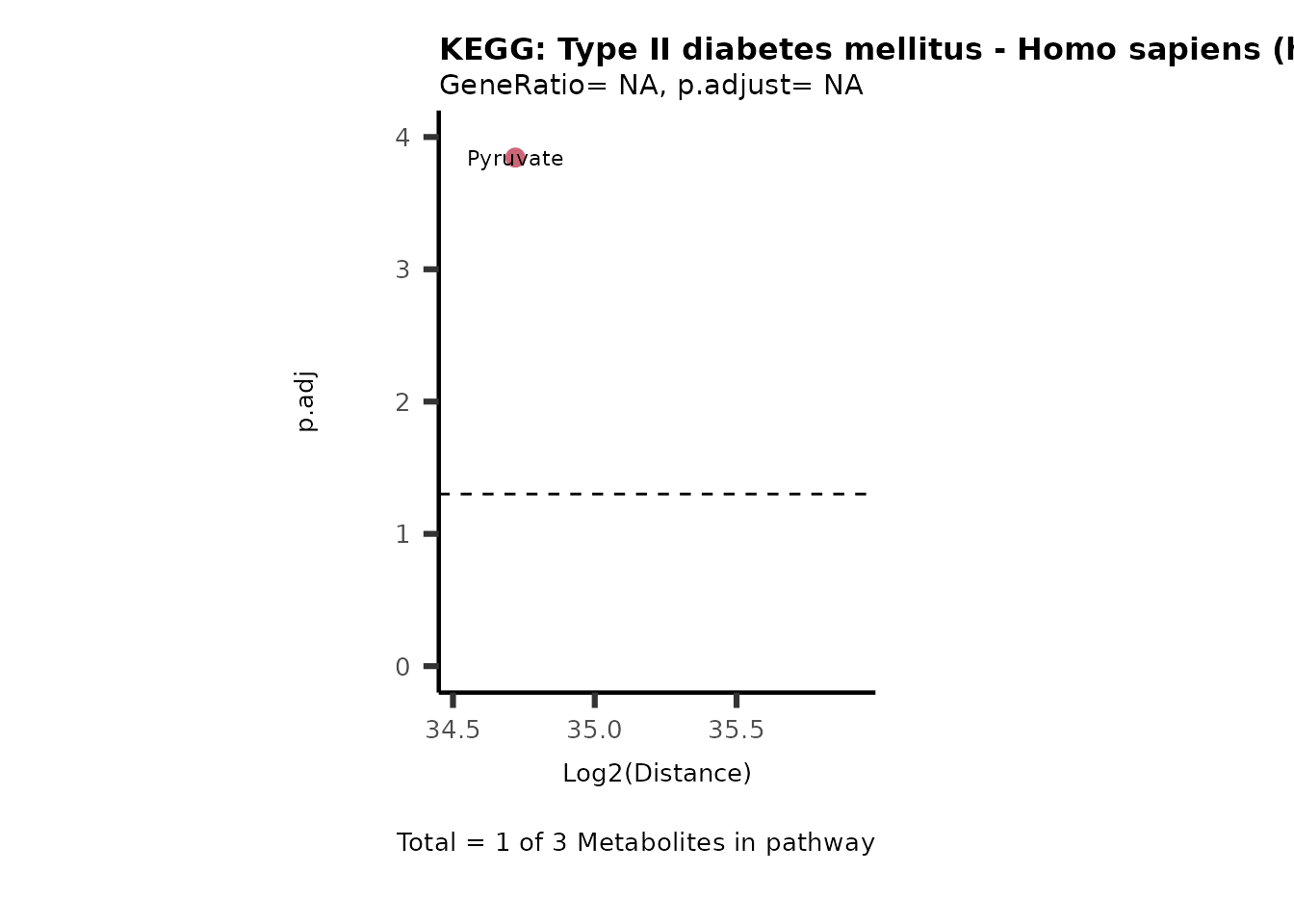
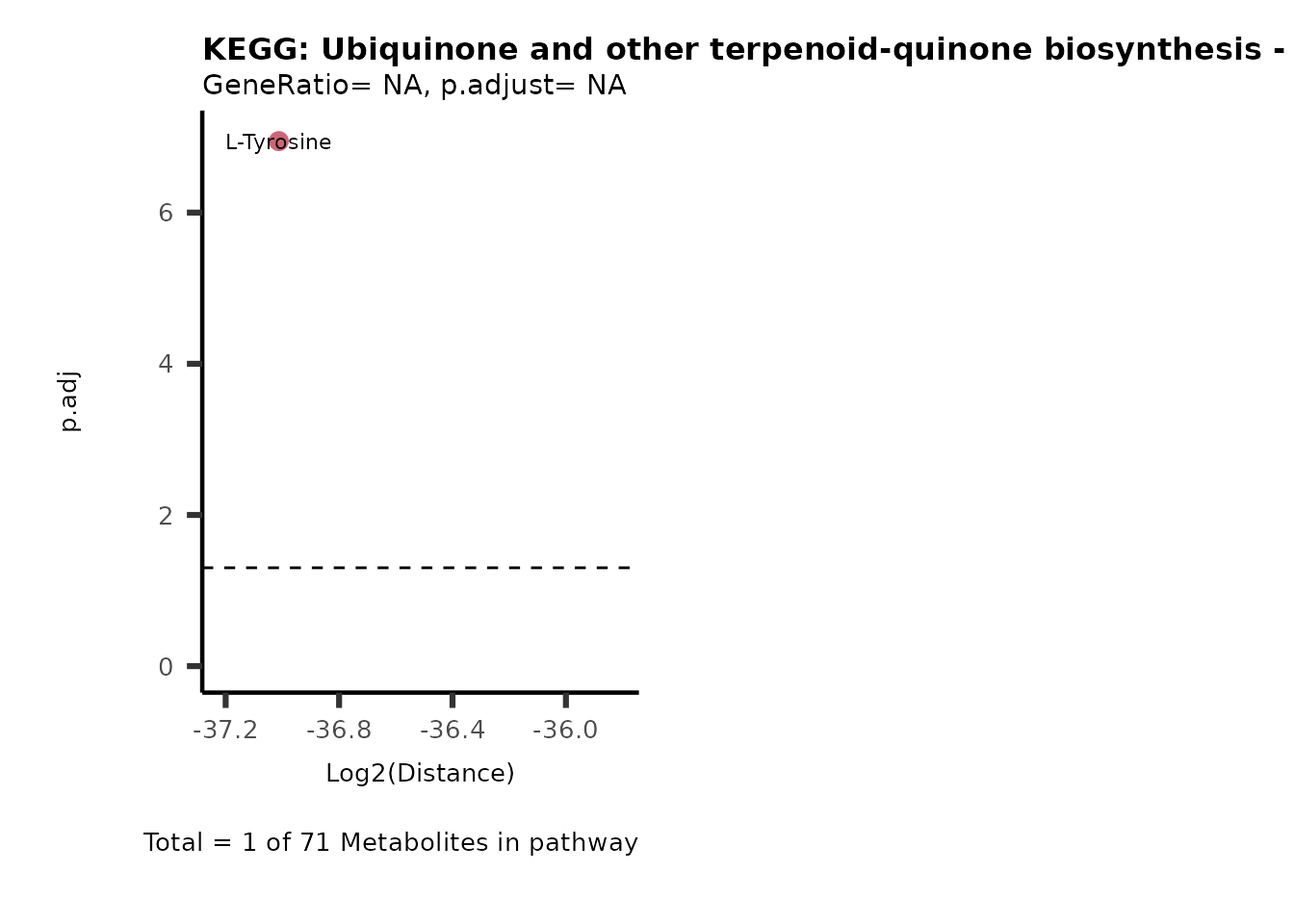
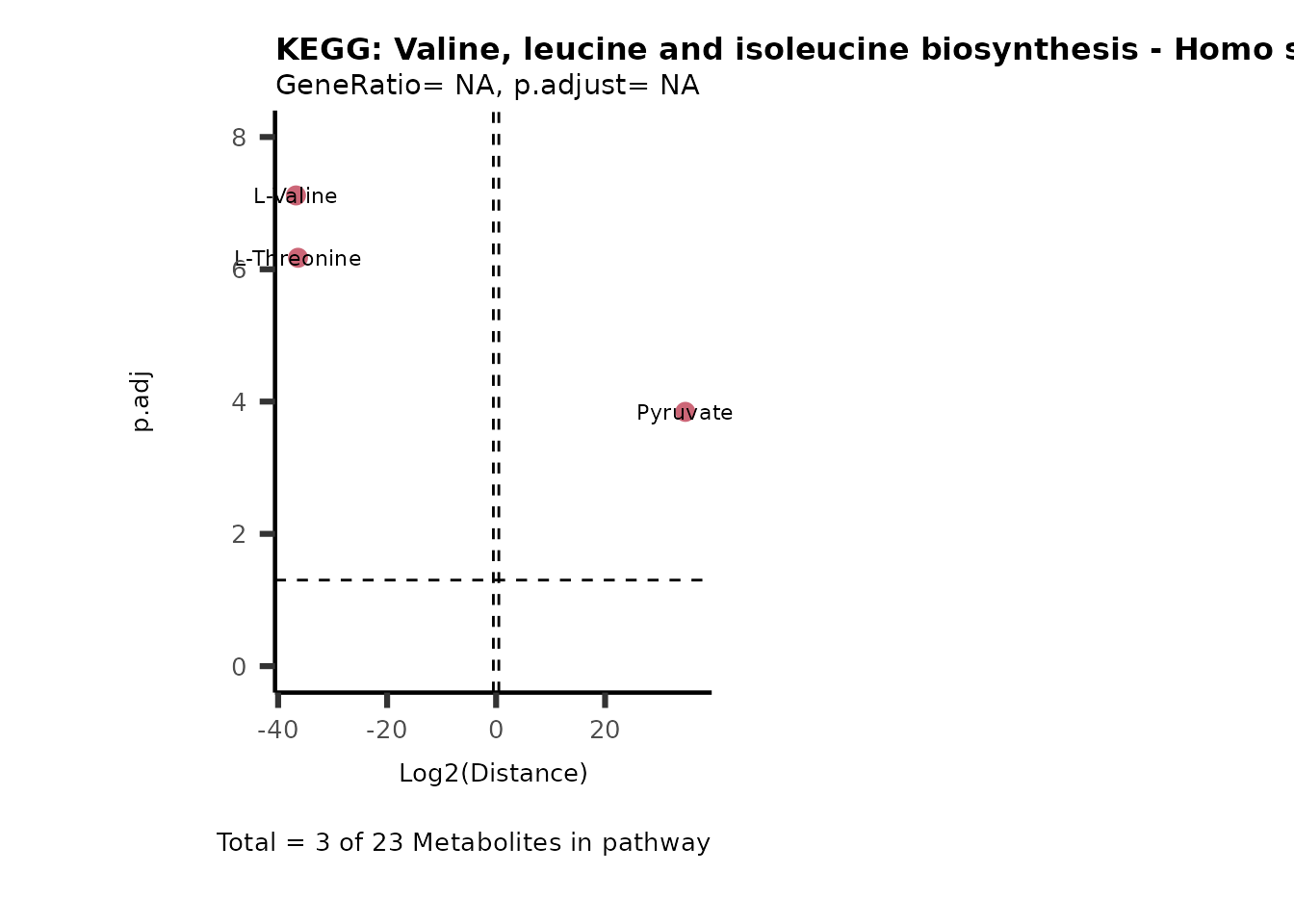
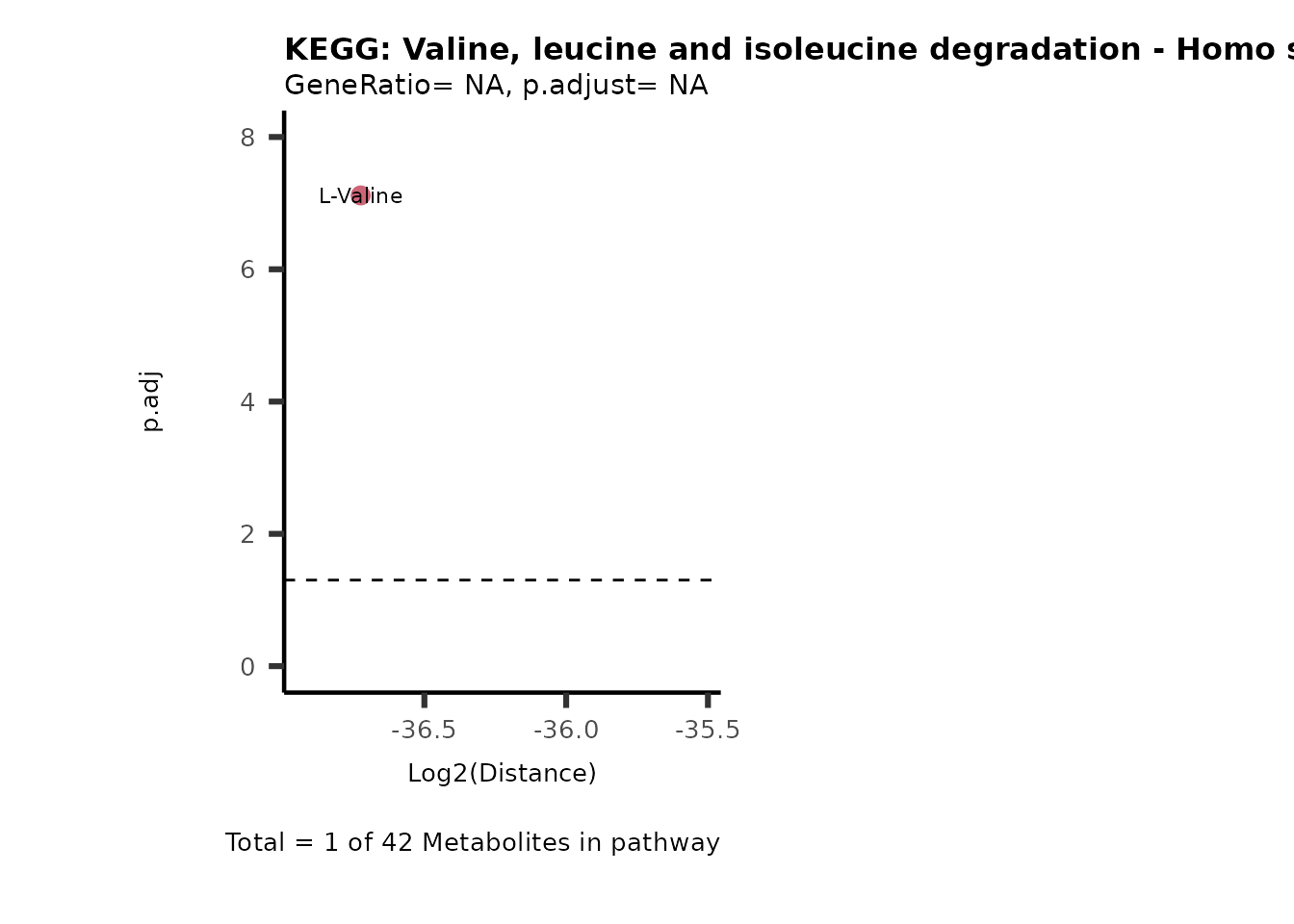
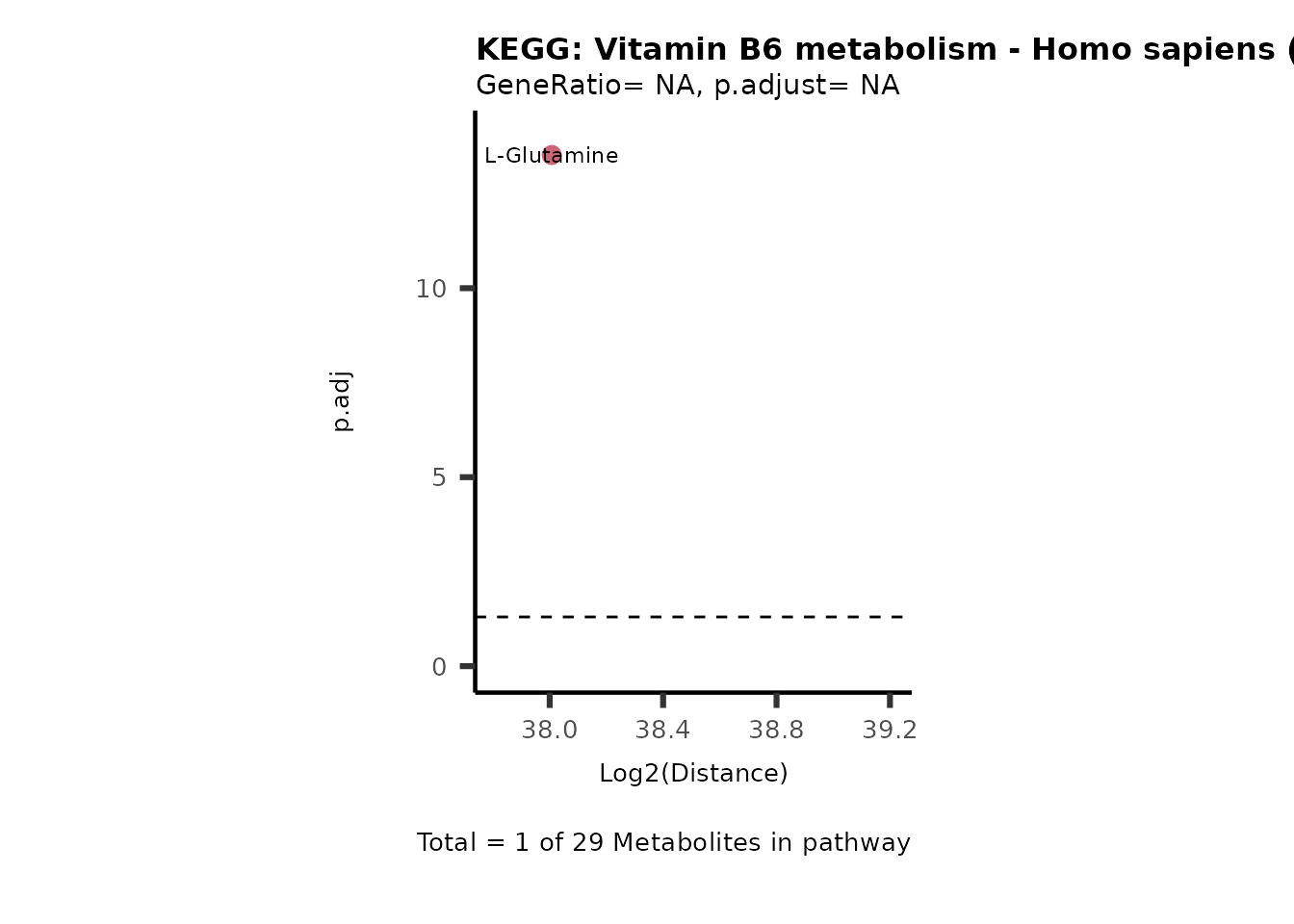
Session information
#> R version 4.4.2 (2024-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.1 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8
#> [6] LC_MESSAGES=C.UTF-8 LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggfortify_0.4.17 rlang_1.1.4 tibble_3.2.1 dplyr_1.1.4 magrittr_2.0.3 MetaProViz_2.1.3
#> [7] ggplot2_3.5.1
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.4.2 later_1.4.1 ggplotify_0.1.2 bitops_1.0-9
#> [5] R.oo_1.27.0 cellranger_1.1.0 polyclip_1.10-7 XML_3.99-0.17
#> [9] factoextra_1.0.7 lifecycle_1.0.4 rstatix_0.7.2 lattice_0.22-6
#> [13] vroom_1.6.5 MASS_7.3-61 backports_1.5.0 limma_3.58.1
#> [17] sass_0.4.9 rmarkdown_2.29 jquerylib_0.1.4 yaml_2.3.10
#> [21] zip_2.3.1 EnhancedVolcano_1.20.0 qcc_2.7 RColorBrewer_1.1-3
#> [25] cowplot_1.1.3 DBI_1.2.3 lubridate_1.9.4 abind_1.4-8
#> [29] zlibbioc_1.48.2 rvest_1.0.4 purrr_1.0.2 R.utils_2.12.3
#> [33] ggraph_2.2.1 BiocGenerics_0.48.1 RCurl_1.98-1.16 hash_2.2.6.3
#> [37] yulab.utils_0.1.8 tweenr_2.0.3 rappdirs_0.3.3 GenomeInfoDbData_1.2.11
#> [41] IRanges_2.36.0 S4Vectors_0.40.2 enrichplot_1.22.0 ggrepel_0.9.6
#> [45] tidytree_0.4.6 pheatmap_1.0.12 pkgdown_2.1.1 svglite_2.1.3
#> [49] codetools_0.2-20 DOSE_3.28.2 xml2_1.3.6 ggforce_0.4.2
#> [53] tidyselect_1.2.1 aplot_0.2.4 farver_2.1.2 viridis_0.6.5
#> [57] stats4_4.4.2 jsonlite_1.8.9 tidygraph_1.3.1 Formula_1.2-5
#> [61] systemfonts_1.1.0 tools_4.4.2 progress_1.2.3 treeio_1.26.0
#> [65] ragg_1.3.3 Rcpp_1.0.13-1 glue_1.8.0 gridExtra_2.3
#> [69] xfun_0.49 qvalue_2.34.0 GenomeInfoDb_1.38.8 withr_3.0.2
#> [73] fastmap_1.2.0 digest_0.6.37 gridGraphics_0.5-1 timechange_0.3.0
#> [77] R6_2.5.1 textshaping_0.4.1 colorspace_2.1-1 GO.db_3.18.0
#> [81] gtools_3.9.5 RSQLite_2.3.9 R.methodsS3_1.8.2 tidyr_1.3.1
#> [85] generics_0.1.3 data.table_1.16.4 graphlayouts_1.2.1 prettyunits_1.2.0
#> [89] httr_1.4.7 htmlwidgets_1.6.4 scatterpie_0.2.4 inflection_1.3.6
#> [93] pkgconfig_2.0.3 gtable_0.3.6 blob_1.2.4 XVector_0.42.0
#> [97] shadowtext_0.1.4 clusterProfiler_4.10.1 OmnipathR_3.15.2 htmltools_0.5.8.1
#> [101] carData_3.0-5 fgsea_1.33.1 scales_1.3.0 kableExtra_1.4.0
#> [105] Biobase_2.62.0 png_0.1-8 ggfun_0.1.8 knitr_1.49
#> [109] rstudioapi_0.17.1 tzdb_0.4.0 reshape2_1.4.4 nlme_3.1-166
#> [113] checkmate_2.3.2 curl_6.0.1 cachem_1.1.0 stringr_1.5.1
#> [117] parallel_4.4.2 vipor_0.4.7 HDO.db_0.99.1 AnnotationDbi_1.64.1
#> [121] desc_1.4.3 pillar_1.10.0 grid_4.4.2 logger_0.4.0
#> [125] vctrs_0.6.5 ggpubr_0.6.0 car_3.1-3 beeswarm_0.4.0
#> [129] evaluate_1.0.1 readr_2.1.5 cli_3.6.3 compiler_4.4.2
#> [133] crayon_1.5.3 ggsignif_0.6.4 labeling_0.4.3 plyr_1.8.9
#> [137] fs_1.6.5 ggbeeswarm_0.7.2 stringi_1.8.4 viridisLite_0.4.2
#> [141] BiocParallel_1.36.0 munsell_0.5.1 Biostrings_2.70.3 lazyeval_0.2.2
#> [145] GOSemSim_2.28.1 Matrix_1.7-1 hms_1.1.3 patchwork_1.3.0
#> [149] bit64_4.5.2 KEGGREST_1.42.0 statmod_1.5.0 igraph_2.1.2
#> [153] broom_1.0.7 memoise_2.0.1 bslib_0.8.0 ggtree_3.10.1
#> [157] fastmatch_1.1-4 bit_4.5.0.1 readxl_1.4.3 gson_0.1.0
#> [161] ape_5.8-1